ГОСТ Р 51352-2013
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
МЕДИЦИНСКИЕ ИЗДЕЛИЯ ДЛЯ ДИАГНОСТИКИ ИН ВИТРО
Методы испытаний
In vitro diagnostic medical devices. Test methods
ОКС 11.020
Дата введения 2015-01-01
Предисловие
1 РАЗРАБОТАН Федеральным государственным бюджетным учреждением здравоохранения "Головной центр гигиены и эпидемиологии" Федерального медико-биологического агентства.
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 380 "Клинические лабораторные исследования и диагностические тест-системы ин витро"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 8 ноября 2013 г. N 1532-ст
4 ВЗАМЕН ГОСТ Р 51352-99
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (gost.ru)
Введение
Настоящий стандарт устанавливает методы испытаний и технические требования к изделиям при оценке их эффективности и безопасности.
По проблемам, затронутом в настоящем стандарте, могут быть применены международные, национальные и региональные законы и правила.
1 Область применения
Настоящий стандарт распространяется на медицинские изделия для диагностики ин витро (далее по тексту - изделия) природного или искусственного происхождения, предназначенные для применения в медицинской практике и используемые в клинико-диагностических лабораториях, выполняющих бактериологические, биохимические, иммунологические, медико-биологические, медико-генетические и другие диагностические ин витро исследования, а также на составные части этих изделий, имеющие функциональное медицинское назначение и изготовляемые отдельно.
Для контрольных материалов, входящих в состав изделия или выпускаемых отдельно с учетом их специфики, необходимо дополнительно учитывать требования ГОСТ Р 53133.3.
На основе данного стандарта допускается разрабатывать методики и проводить испытания изделий для санитарно-эпидемиологических исследований.
Стандарт не распространяется на стандартные образцы и калибраторы, требующие установления типа стандартного образца, используемые при валидации методов и внешней оценки качества лабораторных исследований.
Стандарт не распространяется на диагностические и антитоксические лечебные сыворотки (для ин виво).
Стандарт не распространяется на оборудование для диагностики ин витро (приборы, аппараты, анализаторы, аналитические комплексы, средства измерения и программное обеспечение указанного оборудования).
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ Р 51088-97 Наборы реагентов для клинической лабораторной диагностики. Общие технические условия
ГОСТ Р 52361-2005 Контроль объекта аналитический. Термины и определения
ГОСТ Р 52379-2005 Надлежащая клиническая практика
ГОСТ Р 53022.2-2008 Технологии лабораторные клинические. Требования к качеству клинических лабораторных исследований. Часть 2. Оценка аналитической надежности методов исследования (точность, чувствительность, специфичность)
ГОСТ Р 53022.3 Технологии лабораторные клинические. Требования к качеству клинических лабораторных исследований. Часть 3. Правила оценки клинической информативности лабораторных тестов
ГОСТ Р 53133.3-2008 Технологии лабораторные клинические. Контроль качества клинических лабораторных исследований. Часть 3. Описание материалов для контроля качества клинических лабораторных исследований
ГОСТ Р 53434-2009 Принципы надлежащей лабораторной практики
ГОСТ Р ЕН 12322-2010 Изделия медицинские для диагностики in vitro. Питательные среды для микробиологии. Критерии функциональных характеристик питательных сред
ГОСТ Р ЕН 13612-2010 Оценка функциональных характеристик медицинских изделий для диагностики in vitro
ГОСТ Р ЕН 13641-2010 Устранение или снижение риска инфицирования, связанного с реагентами для диагностики in vitro
ГОСТ Р ИСО 13485-2004 Изделия медицинские. Системы менеджмента качества. Системные требования для целей регулирования
ГОСТ Р ИСО 15193-2007 Изделия медицинские для диагностики in vitro. Измерение величин в пробах биологического происхождения. Описание референтных методик выполнения измерений
ГОСТ Р ИСО 15194-2007 Изделия медицинские для диагностики in vitro. Измерение величин в пробах биологического происхождения. Описание стандартных образцов
ГОСТ Р ИСО 15195-2006 Лаборатории медицинские. Требования к лабораториям референтных измерений
ГОСТ 8.315-97 Государственная система обеспечения единства измерений. Стандартные образцы состава и свойств веществ и материалов. Основные положения
ГОСТ ИСО/МЭК 17025-2009 Общие требования к компетентности испытательных и калибровочных лабораторий
Примечание - При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю "Национальные стандарты", который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя "Национальные стандарты" за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены следующие термины с соответствующими определениями:
3.1 аналит: Компонент пробы, указанный в названии исследуемого свойства или измеряемой величины.
Примечание - В клинической лабораторной диагностике аналиты могут иметь различный характер:
а) физических свойств;
б) химических элементов, ионов, неорганических молекул;
в) органических структур с малой молекулярной массой;
г) макромолекул с известной или приблизительно установленной структурой и специфическими биологическими свойствами;
д) клеток, их структурных элементов или клеточных систем;
е) микроорганизмов (бактерий, вирусов, а также грибов и паразитарных организмов размером до 1 мм), их структуры и свойств.
3.2 биологические жидкости: Жидкости организма человека и их компоненты: кровь, моча, ликвор, лимфа, секреты, сыворотка, плазма, слюна, амниотическая жидкость, эякулят и др.
3.3 биологический материал: Биологические жидкости, ткани и экскреты человека [4].
Примечание -
Биоматериалы человека представляют собой сложные системы смеси различных веществ и клеток, являющихся компонентами, т.е. очерченными частями системы. В аналитике компоненты системы подразделяют на "аналиты", "конкомитанты" и "растворители"; последние два вида компонентов обозначают как "матрицу". Матрица охватывает все компоненты материальной системы, исключая аналит. [ГОСТ Р 53022.2-2008, Часть 2, пункт 3.2] |
3.4 биологическая референтная популяция: Однородная популяция индивидуумов, находящихся в хорошо определяемом состоянии здоровья или болезни.
Примечания
1 Данное определение адаптировано Рабочей группой по глобальной гармонизации [1]*.
________________
* Поз. [1], [3] cм. раздел Библиография. - .
2 В случае если биологический референтный интервал популяции предоставлен изготовителем в инструкции по применению, лаборатория, использующая изделие для диагностики ин витро, ответственна за подтверждение того, что биологическая референтная популяция соответствует популяции, обслуживаемой лабораторией.
3 Определенная группа очевидно здоровых индивидуумов или лиц, находящихся в определенном медицинском состоянии. Понятие позволяет связывать референтный интервал с возрастом, полом и этнической принадлежностью референтной популяции, если требуется.
3.5 диагностика ин витро: Определение наличия и (или) количественного содержания клинически (диагностически) значимых аналитов эндогенной и (или) экзогенной природы, в том числе выделение и идентификация микроорганизмов (бактерий, вирусов, грибов, паразитарных организмов), их токсинов и других продуктов жизнедеятельности в образцах биологических материалов (биологических жидкостей, экскретов, тканей), взятых или выделенных из организма человека.
3.6 идентификация изделия: Установление тождественности характеристик изделия его существенным признакам.
3.7 измерение: Процесс (совокупность операций) экспериментального получения одного или более значений величины.
Примечания
1 Этапы проведения измерения от получения пробы, преобразования аналита в форму удобную для измерения, получения величин в средстве измерения и обработки полученных результатов являются частями процесса измерения. В результате процесса измерения могут быть получены количественные или качественные данные, представляющие значимую для диагностики информацию.
2 Качественные данные - результат, полученный дихотомическим делением по определенному признаку.
Пример - данные ИФА на наличие инфицированности пациента определяется по признаку ОП пробы больше или меньше ОП критического в данной системе.
3 К измерению не относится визуальное исследование свойств, например цвета.
4 Федеральные органы исполнительной власти, осуществляющие нормативно-правовое регулирование в областях деятельности (здравоохранения), определяют измерения, относящиеся к сфере государственного регулирования обеспечения единства измерений, и устанавливают к ним обязательные метрологические требования, в том числе показатели точности измерений. [2]
3.8 интерсепт: Концентрация анализируемого вещества, которая соответствует определенной точке на калибровочном графике.
3.9 контрольный материал: Однородный материал человеческого или животного происхождения или искусственный материал, приближающийся насколько это возможно, по своим наиболее существенным свойствам к исследуемому биологическому материалу пробы и предназначенный для оценки прецизионности исследований проб пациентов, выполняемых в клинико-диагностических лабораториях.
Примечание - К контрольным материалам относят контрольные лабораторные растворы с определенной концентрацией чистого анализируемого вещества (в случае с ферментами предпочтительнее сыворотка или плазма крови человека с известной активностью фермента).
3.10 коэффициент вариации: Показатель воспроизводимости результатов определения, рассчитанный как отношение значения среднего квадратического отклонения к среднему арифметическому значению.
3.11 линейность: Отклонение значения концентрации определяемого вещества (или активности фермента) от теоретического в диапазоне рабочих концентраций.
3.12 медицинское изделие для диагностики ин витро: Любое медицинское изделие, предназначенное изготовителем для исследования без контакта с пациентом образцов биологического материала человека при отдельном применении или в комбинации с другими медицинскими изделиями исключительно с целью получения данных относительно физиологического или патологического состояния, и (или) относительно проблем внутриутробного развития плода, и (или) для мониторинга терапевтических мероприятий, и (или) для определения совместимости тканей.
Примечания
1 Данное определение адаптировано Рабочей группой по глобальной гармонизации [1].
2 Может быть использовано отдельно или в комбинации с принадлежностями или другими медицинскими изделиями.
3 Медицинские изделия для диагностики ин витро включают в себя реагенты, калибраторы, контрольные материалы. Медицинские изделия для диагностики ин витро могут включать емкости для сбора и хранения образцов, программное обеспечение и связанные с ним инструменты, приборы или другие предметы, используемые для целей диагностики или в помощь диагностике, но не рассматриваемые в данном стандарте.
3.13 метод: Общее описание логической последовательности операций при проведении исследований, измерений для диагностики ин витро.
Примечания
1 Основными методами в клинико-лабораторной практике являются:
а) Радиоиммунологический - метод, основанный на иммунохимической реакции, проводимой ин витро в присутствии меченного радионуклидом антигена или антитела.
б) Иммуноферментный - метод, основанный на иммунохимической реакции, проводимой ин витро в присутствии меченого ферментом соединения и ферментзависимого субстрата.
в) Иммунофлюоресцентный - метод, основанный на иммунохимической реакции, проводимой ин витро в присутствии флуоресцентной метки.
г) Иммунохемилюминесцентный - метод, основанный на иммунохимической реакции, проводимой ин витро в присутствии хемилюминесцентной метки.
д) Иммунохроматографический - метод, основанный на конкуренции определяемого вещества и конъюгата аналога определяемого вещества за субстрат на полоске хроматографической бумаги.
е) Микроанализ нуклеотидных последовательностей - метод, основанный или на процессе специфического умножения количества исследуемых участков дезоксирибонуклеиновой кислоты (ДНК) или рибонуклеиновой кислоты (РНК), в том числе методом полимеразной цепной реакции (ПЦР) с последующей детекцией сигнала, или на прямой детекции сигналов, возникающих при гибридизации с синтетическими зондами.
ж) Фотометрический - метод, основанный на избирательном поглощении инфракрасного, видимого или ультрафиолетового излучения молекулами определяемого вещества или его соединения с соответствующим реагентом.
з) Коагулометрический - способ оценки системы свертывания крови, основанный на регистрации времени фибринообразования.
и) Микробиологические методы - "золотой стандарт" микробиологической диагностики, позволяют точно установить факт наличия возбудителя, идентификацию (до вида микроорганизма) с учётом морфологических, тинкториальных, культуральных, биохимических, токситенных и антигенных свойств микроорганизма и определение чувствительности к антимикробным препаратам у выделенного возбудителя.
к) Биофизический - метод, характеризующий свойства биологической жидкости, например, вязкость, температуру замерзания и т.д.
2 Метод может быть основой одной или нескольких методик измерений, каждая из которых имеет присущие ей числовые значения характеристик выполнения.
3.14 методика измерений: Детальное описание измерений в соответствии с одним или более методом.
Примечания
1 Методику измерений обычно описывают достаточно подробно и представляют в виде документа (инструкции), позволяющей пользователю выполнить измерение.
2 Методика измерения может включать информацию о неопределенности измерений.
3 В ряде случаев методика измерений, прописанная достаточно детально и способная обеспечить пользователя всей необходимой информацией без привлечения дополнительных источников, может рассматриваться как стандартная операционная процедура.
3.15 микробиологические питательные среды (ПС): Медицинские изделия, предназначенные для выявления, накопления, культивирования, дифференциальной диагностики, транспортировки и хранения микроорганизмов.
3.16 оценка соответствия: Доказательство того, что заданные требования к изделию выполнены.
3.17 оценка функциональной характеристики (performance evaluation): Изучение медицинского изделия для диагностики ин витро с целью установления или проверки заявленной функциональной характеристики.
3.18 полимеразная цепная реакция (ПЦР): Экспериментальный метод молекулярной биологии, позволяющий добиться значительного увеличения малых концентраций определенных фрагментов нуклеиновой кислоты (ДНК) в биологическом материале (пробе). Помимо амплификации ДНК, ПЦР позволяет производить множество других манипуляций с нуклеиновыми кислотами (введение мутаций, сращивание фрагментов ДНК) и широко используется в биологической и медицинской практике, например, для диагностики заболеваний (наследственных, инфекционных), для установления отцовства, для клонирования генов, выделения новых генов.
3.19 специфичность: Способность изделия определять только то вещество (или активность фермента, или участок генома), для которого изделие предназначено.
3.20 стандартный образец (аттестованный стандартный образец): Стандартный образец, с сопроводительной документацией, выданной авторитетным органом, (уполномоченным) и основанной на подтвержденных процедурах, использованных для получения значения специфического свойства с неопределенностью и прослеживаемостью.
Пример - Сыворотка человека, содержащая холестерин с приписанным значением величины и неопределенностью измерения, утвержденной приложенным сертификатом, предназначенная для использования как калибратор или контрольный материал правильности.
Примечания
1 Данное определение адаптировано Рабочей группой по глобальной гармонизации [1].
2 В этом определении "неопределенность" охватывает и "неопределенность измерения" и "неопределенность качественного свойства", такого как идентичность и последовательность, выраженных как вероятности. Прослеживаемость означает "метрологическую прослеживаемость" значения величины и прослеживаемость номинального свойства.
3 Адаптировано из ИСО/МЭК Руководство 99:2007, пункт 5.14 [3].
3.21 таксон (лат. taxon): Группа живых организмов, объединённых на основании определенных методов классификации.
3.22 тест на "открытие" (метод добавок): Проверка соответствия значения определяемой концентрации вещества (или активности фермента) расчетной величине, полученной путем смешивания равных объемов контрольных растворов (калибровочных проб, контрольных сывороток или биологического материала) с установленной концентрацией (или активностью).
4 Общие требования к испытаниям медицинских изделий для диагностики ин витро
4.1 Виды испытаний
4.1.1 Изделия подвергаются следующим видам испытаний:
а) предварительные;
б) технические;
в) клинические;
г) государственный контроль качества;
д) сертификационные.
Примечание - Токсикологические испытания изделий, в связи с их неактуальностью для диагностики ин витро, в данном стандарте не рассматриваются. Устранение или снижение риска инфицирования, связанного с содержанием материала человеческого, животного происхождения или полученного в биотехнологическом процессе, когда результаты анализа выявляют возможность заражения человека, проводится с учетом требований по ГОСТ Р ЕН 13641 и действующих санитарных требований.
4.1.2 Предварительные испытания проводятся изготовителем с учетом требований ГОСТ Р ИСО 13485.
4.1.3 Технические и сертификационные испытания изделий проводят в испытательных центрах (лабораториях), аккредитованных и признанных независимыми в установленном законодательством порядке.
4.1.4 Клинические испытания изделий для диагностики ин витро проводятся в медицинских и научных учреждениях, имеющих лицензии на соответствующие виды деятельности. Испытания проводятся с использованием биологического материала без участия человека.
4.1.5 Государственный контроль качества включает проверку изделий на соответствие всем требованиям нормативной и технической документации. Государственный контроль за выпускаемой продукцией включает предварительный, последующий и, при необходимости, арбитражный контроль качества изделий и проводится соответствующими службами Минздрава России. Порядок проведения государственного контроля, периодичность и минимальный объем выборки по каждому конкретному классу изделий устанавливаются соответствующими нормативными документами, утвержденными в установленном порядке.
4.1.6 Сертификационным испытаниям подвергают изделия, прошедшие приемочные, технические и клинические испытания, разрешенные к производству и применению в медицинской практике и зарегистрированные в установленном порядке.
4.2 Общие требования к лабораториям при проведении испытаний
4.2.1 Испытания по оценке соответствия (далее - оценка соответствия) изделия проводят с целью государственной регистрации, государственного контроля или сертификации. При оценке соответствия учитывается, отвечает ли изделие требованиям технических условий, действующих нормативных документов, сводов правил (например, санитарных правил и норм, других подобных документов).
4.2.2 Оценка соответствия путем технических испытаний или сертификации проводится в испытательных лабораториях, аккредитованных по требованиям ГОСТ ИСО/МЭК 17025 в установленном законом порядке. Высокоспециализированные лаборатории (калибровочные, референтных измерений), проводящие испытания медицинских изделий для диагностики ин витро, используемые в лабораторной медицине, должны соответствовать специальным требованиям, установленным в ГОСТ Р ИСО 15195.
Примечания -
1
Лаборатории референтных измерений должны обеспечивать прослеживаемость до наиболее высокого достижимого метрологического уровня по сравнению с уровнем прослеживаемости, установленным для лабораторий рутинных измерений. Метрологический уровень результатов, получаемых в лабораториях референтных измерений, должен содействовать тому, чтобы лаборатории рутинных измерений соответствовали медицинским требованиям. [ГОСТ Р ИСО 15195-2006, Введение]. |
2
Эти высокоспециализированные лаборатории могут быть связаны или работать по контракту с такими организациями, как национальные метрологические институты, организации, осуществляющие внешнюю оценку качества исследований, научные центры, производители медицинских изделий для диагностики in vitro. Лаборатории референтных измерений должны внедрять методики референтных измерений и получать результаты, которые являются правильными и прослеживаемыми до национальных или международных первичных стандартных образцов, если таковые доступны. При наличии такой возможности прослеживаемость должна быть установлена по стандартным образцам, свойства которых выражены в единицах Международной системы единиц СИ (ИСО 17511:2003). Во многих случаях свойства биологических материалов не могут быть выражены в единицах СИ, поскольку молекулярная структура этих аналитов недостаточно изучена и возможны отличия стандартного образца от того аналита, что находится в нативной пробе человеческого происхождения (например, состояние гликозилирования белка); тогда цепь прослеживаемости заканчивается на нижнем уровне, например на произвольной международной единице. Однако лаборатории референтных измерений должны представлять значения стандартных образцов, полученных от заказчиков, прослеженные до наивысшего возможного уровня методик референтных измерений или стандартных образцов. Даже если значение свойства биологического материала не прослежено до единицы СИ, на каждом этапе процедуры референтного измерения (например, гравиметрии, волюметрии, измерения температуры) должны быть получены значения, которые прослежены до соответствующей единицы СИ. Концепция прослеживаемости, ее применения и ограничения детально описаны в ИСО 17511:2003. [ГОСТ Р ИСО 15195-2006, Введение] |
4.2.3 Разработка программы проведения технических и клинических испытаний должна проводится с учетом ГОСТ Р ЕН 13612. В программе проведения технических и клинических испытаний необходимо учитывать требования к качеству клинических исследований по оценке аналитической надежности методов исследования (точность, чувствительность, специфичность) по ГОСТ Р 53022.2 и по оценке клинической информативности по ГОСТ Р 53022.3.
4.2.4 Медицинские учреждения, проводящие клинические испытания медицинских изделий для диагностики ин витро, должны соответствовать требованиям по надлежащей лабораторной практике ГОСТ Р 53434, в случаях привлечения человека как объекта при проведении испытаний, соответствовать требованиям ГОСТ Р 52379.
4.2.5 Описание методик референтных измерений и описание стандартных образцов должно соответствовать требованиям ГОСТ Р ИСО 15193 и ГОСТ Р ИСО 15194. Оценка соответствия изделий и оценка функциональных характеристик изделий в форме испытаний проводится с учетом настоящего стандарта и всей действующей нормативной и правовой документации.
4.2.6 Результатом технических, клинических испытаний медицинских изделий для диагностики ин витро с учетом классификации в зависимости от потенциального риска их применения, и экспертизы качества, эффективности и безопасности медицинских изделий является заключение о полноте результатов проведенных испытаний и исследований. Заключение должно подтвердить, что в результате проведенных испытаний доказано:
а) функциональные и технические характеристики соответствуют заявленным производителем в технической и эксплуатационной документации, а также выполнены обязательные требования нормативной и правовой документации;
б) эффективность изделия подтверждена, функциональные характеристики изделия позволяют исследовать аналит (или его активность). Искомый аналит находится в причинно-следственной связи с предполагаемой патологией, и что доказана полезность данного теста для диагностики и выбора лечебной стратегии.
в) изготовитель (производитель) предпринял все меры для максимального снижения потенциального и косвенного риска применения изделия и условий, в которых данное изделие предназначено для применения, учел квалификацию и опыт предполагаемых пользователей, и, где применимо, физическое состояние пользователей с ограниченными возможностями.
5 Оценка соответствия медицинского изделия
5.1 Идентификация изделия
Идентификация изделия проводится по требованиям ГОСТ Р 51088, технической и эксплуатационной документации производителя и его маркировке.
5.1.1 Проверка комплектности и составных частей изделия:
а) комплектность в соответствии с эксплуатационной документацией;
б) внешний вид и состояние упаковки изделия и его компонентов;
в) наличие маркировки изделия и его компонентов, подтверждающей соответствие требованиям нормативной документации изготовителя.
5.1.2 Компоненты должны быть подвергнуты визуальному контролю.
Осмотр компонентов изделия заключается в установлении соответствия показателей внешнего вида требованиям технической и эксплуатационной документации:
а) агрегатное состояние (таблетка, порошок, аморфная масса, жидкость и т.п.);
б) цвет;
в) прозрачность (для жидкостей);
г) наличие или отсутствие осадка или хлопьев (для жидкостей);
д) консистенция.
5.2 Общие технические требования к медицинским изделиям для диагностики ин витро при проведении оценки соответствия
5.2.3 Общие требования
5.2.3.1 Оценка эффективности и безопасности должна осуществляться при непосредственном сопоставлении с данными, полученными при проведении референтной методики на аттестованных современных изделиях. Если используемое для сравнения изделие в момент проведения сопоставления показателей представлено на рынке, оно должно быть зарегистрировано в установленном порядке.
5.2.3.2 Если в ходе проведения оценки получаются расходящиеся результаты тестирования, такие расхождения должны быть по возможности устранены, например:
а) посредством оценки отличающейся пробы в других тестовых системах;
б) за счет использования референтного метода или маркера;
в) посредством проверки клинического состояния и диагноза пациента;
г) в ходе тестирования повторных контрольных образцов.
5.2.3.3 Оценка эффективности проводится на пробах, полученных от биологической референтной популяции.
5.2.3.4 Положительные образцы, использованные при оценке эффективности, должны выбираться так, чтобы отражать различные стадии соответствующего заболевания (заболеваний), разный спектр антител, различные генотипы, различные субтипы, мутации и пр.
5.2.3.5 Отрицательные пробы, используемые для оценки эффективности, подбираются таким образом, чтобы достоверно отражать население, для которого предназначается тест, например, доноров крови, госпитализированных пациентов, беременных женщин и пр.
5.2.3.6 Изделия должны обладать специфичностью не ниже клинически обоснованной диагностической эффективности, если производителем не обоснованы другие требования. Специфичность рассчитывается на основании частоты повторно реактивных (ложноположительных) результатов для проб с отрицательной реакцией на целевой маркер.
Пример - Медицинские изделия, предназначенные для определения групп крови, должны обладать специфичностью по образцам донорской крови не менее 99,5%.
5.2.3.7 Для доказательства эффективности изделия производитель должен в полном объеме провести проверку, с тем чтобы выявить эффект потенциально интерферирующих веществ. Оцениваемые потенциально интерферирующие вещества будут в определенной степени зависеть от состава реагента и конфигурации анализа. Потенциально интерферирующие вещества определяются в ходе анализа рисков, обязательного для соблюдения основных требований к каждому новому изделию, но могут также включать, например:
а) образцы, представляющие "родственные" инфекции;
б) пробы, взятые у многорожавших женщин, то есть женщин, у которых было более одной беременности, или пациентов с положительным ревматоидным фактором;
в) в случае рекомбинантных антигенов - антитела человека к компонентам системы экспрессии, например, антитела к Е. coli или антитела к дрожжам.
5.2.3.8 В рамках обязательного анализа рисков в ходе повторного анализа слабоположительных образцов определяется общая частота ошибок системы, приводящих к ложноотрицательным результатам.
5.2.3.9 Если в отношении нового медицинского изделия для диагностики ин витро, отнесенного к высокому классу, специально не оговорены общие технические требования, то следует принимать во внимание требования для изделия, например, со сходным применением или со сходными рисками применения.
5.2.4 Изделия для выявления вирусных инфекций
5.2.4.1 Предлагаемые для обращения на рынке в качестве скрининговых или диагностических тестов изделия для выявления вирусных инфекций должны удовлетворять требованиям к чувствительности и специфичности. Для оценки эффективности скрининговых тестов исследуются образцы, полученные из различных (не менее двух) источников, включающие образцы последовательной сдачи крови.
5.2.4.2 Оценка эффективности тестов для скрининга должна включать 25 положительных (если есть в наличии в случае редких инфекций) "взятых в тот же день" свежих проб сыворотки и/или плазмы (не более 1 дня после отбора пробы).
5.2.5 Изделия, предназначенные для тестирования жидкостей организма
5.2.5.1 Изделия, предназначенные для диагностики биологических агентов, мочи, слюны и пр., должны соответствовать таким же требованиям в отношении чувствительности и специфичности, что и тесты для сыворотки или плазмы. Для оценки эффективности проводится тестирование проб от тех же лиц в обоих тестах с подтверждением по анализу соответствующей пробы сыворотки или плазмы.
5.2.6 Медицинские изделия, предназначенные для самодиагностики
5.2.6.1 К изделиям, предназначенным для самодиагностики, то есть для домашнего использования, должны предъявляться те же требования к оценке функциональных характеристик (чувствительности и специфичности), что и к изделиям для профессионального использования.
5.2.6.2 Наиболее важные этапы оценки эффективности должны проводиться (или повторяться) соответствующими пользователями-непрофессионалами, чтобы проверить работоспособность изделия и инструкцию по его использованию.
5.2.7 Оценка чувствительности по положительным результатам испытаний аттестованных положительных образцов сероконверсии
5.2.7.1 Чувствительность диагностического теста в ходе сероконверсии должна соответствовать современным требованиям. Если дальнейшее тестирование с использованием той же или дополнительной панели сероконверсии проводит испытательный орган, результаты должны подтверждать первоначальные данные оценки эффективности. Панели сероконверсии должны начинаться с отрицательной пробы (проб) крови и иметь минимальный временной интервал между ее заборами.
5.2.7.2 Все положительные образцы должны подтверждаться как положительные с помощью испытуемого изделия. Новое изделие должно иметь функциональные характеристики, как минимум, эквивалентные показателям современного ранее апробированного изделия.
5.2.7.3 Для изделий, выявляющих антитела к вирусам иммунодефицита человека, необходимо проанализировать как минимум 40 образцов ранней сероконверсии ВИЧ. Результаты должны соответствовать современным требованиям.
5.2.8 Оценка эффективности изделия для анализа сыворотки и плазмы
5.2.8.1 Оценка эффективности изделий, предназначенных для анализа сыворотки и плазмы, должна демонстрировать эквивалентность сыворотки к плазме. Это необходимо дополнительно продемонстрировать, по крайней мере, на 50 пробах (25 положительных и 25 отрицательных).
5.2.8.2 Оценка эффективности изделий, предназначенных для анализа плазмы, должна подтвердить показания изделия с использованием всех антикоагулянтов, возможность применения которых с данным изделием указана производителем. Это необходимо продемонстрировать, по крайней мере, на 50 пробах (25 положительных и 25 отрицательных) для каждого антикоагулянта.
5.2.9 Оценки эффективности реагентов для определения антигенов групп крови
5.2.9.1 Оценка эффективности реагентов для определения антигенов групп крови:
- система групп крови ABO АВО1 (А), АВО2 (В), АВО3 (А, В);
- система групп крови Rh RH1 (D), RH2 (С), RH3 (Е), RH4 (с), RH5 (е);
- система групп крови Келла KEL1 (K)
осуществляется при непосредственном сопоставлении с показателями зарегистрированного апробированного современного изделия.
5.2.9.2 Если в ходе проведения оценки результаты тестирования расходятся, такие расхождения должны быть по возможности устранены, например:
а) посредством оценки отличающейся пробы в других тестовых системах;
б) за счет использования альтернативного метода.
5.2.9.3 Положительные образцы, использованные для оценки эффективности, должны выбираться таким образом, чтобы отражать варьирующую и слабую экспрессию антигенов.
5.2.9.4 В рамках оценки эффективности изделия (наборы реагентов) должны проходить проверку, с тем чтобы выявить эффект потенциально интерферирующих веществ. Набор оцениваемых потенциально интерферирующих веществ будет в определенной степени зависеть от состава реагента и структуры анализа. Потенциально интерферирующие вещества определяются в ходе анализа рисков, обязательного для соблюдения основных требований для каждого нового изделия.
5.2.10 ПЦР анализ
5.2.10.1 Результаты количественного анализа ПЦР должны соответствовать требованиям международных стандартов или калиброванным стандартным материалам, если таковые имеются в наличии, и применяться в международных единицах величин, используемых в конкретной области применения.
5.2.10.2 ПЦР анализ может использоваться для обнаружения вируса в образцах отрицательных по антителам, то есть образцах до сероконверсии. Вирусы, связанные в иммунные комплексы, могут вести себя иначе по сравнению со свободными вирусами, например, на стадии центрифугирования. Поэтому важно при изучении устойчивости включать в рассмотрение образцы, отрицательные по антителам (до сероконверсии).
5.2.10.3 Для исследования возможной кросс-контаминации в ходе изучения устойчивости необходимо провести, по крайней мере, пять циклов анализа с попеременно меняющимися высокотитражными и отрицательными образцами. Высокотитражные образцы должны включать пробы со встречающимися в естественных условиях высокими титрами вируса.
5.3 Испытания изделий
5.3.1 Экспериментальная проверка изделий должна быть основана на контроле качества системы, включая методику определения искомого аналита, контрольных материалов, изделия конкретного вида.
5.3.2 Средства измерения, дозирующие устройства, весы и другое оборудование, используемые при испытаниях, должны быть поверены в соответствии с требованиями, предъявляемыми к средствам измерения.
5.3.3 Высокотехнологичное лабораторное оборудование, используемое при проведении испытания, должно находиться в исправном состоянии с документально подтвержденным техническим обслуживанием.
5.3.4 Результатом проведенных испытаний является Акт с протоколами лабораторных (технических) испытаний, подтверждающие соответствие или несоответствие изделия и методики проведения измерения заявленным функциональным характеристикам.
5.3.5 Образцы изделий представляют на испытания в количестве, необходимом для проведения трех полноценных видов испытаний (межсерийного, внутрисерийного испытания) с учётом необходимого объема исследований образцов в соответствии с требованиями нормативных документов.
5.3.6 Проведение испытаний должно соответствовать согласованной программе испытаний.
5.4 Оценка показателей функционирования изделий для различных методов анализа
5.4.1 Изделия для радиоиммунологического анализа
5.4.1.1 Общая радиоактивность меченого компонента
Значение общей радиоактивности компонента, меченого радионуклидом ( или
), является основанием для расчета некоторых других показателей. Кроме того, определение общей радиоактивности позволяет проконтролировать количество поставляемого меченого вещества.
Радиоактивность компонента А, килобеккерели (кБк), меченого , определяют на день паспортизации набора по формуле:
![]() , (1)
, (1)
где - скорость счета пробы меченого компонента, имп/мин;
- общий объем меченого компонента в наборе, см
;
- объем пробы меченого компонента, взятой для измерения скорости счета;
- эффективность счета гамма-счетчика, доли единицы;
![]() - коэффициент перевода импульсов в минуту в килобеккерели.
- коэффициент перевода импульсов в минуту в килобеккерели.
Радиоактивность компонента , кБк, меченого ЗН, определяют на день паспортизации изделия по формуле:
![]() , (2)
, (2)
где - скорость счета пробы меченого компонента, имп/мин;
- общий объем меченого компонента в наборе, см
;
- объем пробы меченого компонента, взятой для измерения скорости счета;
- эффективность счета гамма-счетчика, доли единицы;
![]() - коэффициент перевода импульсов в минуту в килобеккерели;
- коэффициент перевода импульсов в минуту в килобеккерели;
- коэффициент поправки на гашение (определяют экспериментально путем измерения скорости счета образцового радиоактивного раствора трития в соответствующей сцинтилляционной жидкости до и после внесения в нее определенного количества растворенного меченого компонента и компонента, не содержащего меченого соединения).
5.4.1.2 Тотальная (общая) радиоактивность, внесенная в анализируемую пробирку
Для расчета ряда показателей качества изделия необходимо определить тотальную (общую) радиоактивность , внесенную в анализируемую пробирку. Значение
определяется как числовой результат скорости счета радиоактивности, внесенной в каждую аналитическую пробирку.
Для определения в пробирки вносят компонент, содержащий радионуклид, в объеме, указанном в инструкции по применению изделия, после чего добавляют в пробирки все компоненты инкубационной среды в объеме, указанном в инструкции по применению изделия; центрифугирование не производят.
Радиометрию каждой пробирки проводят в отдельности и определяют среднее арифметическое значение и значение коэффициента вариации. Достоверность сосчитанных импульсов должна быть в пределах от 1% до 2%, коэффициент вариации счета проб не должен превышать 3%, интенсивность счета
должна быть не менее 10000 имп/мин для получения достоверности счета с погрешностью менее 1%.
5.4.1.3 Специфически связанная радиоактивность (специфическое связывание)
Специфически связанную радиоактивность (или специфическое связывание) определяют с помощью нулевой калибровочной пробы, то есть при нулевой концентрации определяемого вещества.
Значение специфического связывания (применяют также термин "максимальное связывание") выражают в процентах как отношение значения радиоактивности образца с нулевой концентрацией, из которого исключено значение неспецифически связанной радиоактивности, к общей радиоактивности образца:
![]() , (3)
, (3)
где - скорость счета в пробирке, содержащей нулевую калибровочную пробу, имп/мин;
- скорость счета тотальной радиоактивности, внесенной в аналитическую пробирку, имп/мин.
Значение специфического связывания дает основную информацию о протекании иммунохимической реакции между антителами и меченым антигеном в данной системе для радиоиммунологического анализа. Его изменения могут быть обусловлены, прежде всего, изменениями концентраций обоих компонентов в реакционной смеси и их свойств, особенно сродства и емкости связи антител, удельной активности радиоиндикатора и его повреждением, а также изменениями, происходящими во время хранения или при инкубации. Кроме того, на значение специфического связывания могут оказать влияние и другие компоненты реакционной смеси, условия инкубации, а также правильность выполнения процедур сепарации. Любое заметное отклонение значения ![]() (более чем на 5%) от обычно получаемых значений при данной методике должно служить сигналом о нарушении стандартных условий определения и качества компонентов изделия, а также должно послужить основанием к дополнительной проверке изделия.
(более чем на 5%) от обычно получаемых значений при данной методике должно служить сигналом о нарушении стандартных условий определения и качества компонентов изделия, а также должно послужить основанием к дополнительной проверке изделия.
Значение специфического связывания определяют следующим образом: в пробирки (от 8 до 10 шт.) вносят нулевую калибровочную пробу и все компоненты реакционной смеси, после чего проводят все процедуры, предусмотренные инструкцией по применению изделия.
В идеале значение специфического связывания должно быть близко к 50%. Чем ниже этот показатель, тем меньше разрешающая способность калибровочной кривой изделия, поскольку калибровочная кривая будет более пологой. При высоком значении ![]() чувствительность изделия увеличивается, но "рабочий" отрезок калибровочной кривой уменьшается.
чувствительность изделия увеличивается, но "рабочий" отрезок калибровочной кривой уменьшается.
5.4.1.4 Неспецифически связанная радиоактивность (неспецифическое связывание)
Неспецифическое связывание - это неспецифически связанная радиоактивность в образце при отсутствии специфических антител.
Определение значения неспецифического связывания дает, прежде всего, информацию об эффективности используемого метода сепарации свободной и связанной фракций и о неспецифическом захвате свободного радиоиндикатора в связанной фракции. Причиной неспецифического связывания, прежде всего, является неспецифическая сорбция свободного радиоиндикатора на стенках пробирок, на поверхности осадка и сорбентов. Кроме того, неспецифическое связывание - это влияние перекрестных реакций в анализируемых образцах в присутствии эндогенных антител и воздействие некоторых неспецифических факторов вследствие их присутствия в анализируемых образцах.
Поскольку неспецифическое связывание - одна из основных проблем радиоиммунологического анализа, его исследование и контроль важны и необходимы. При этом значение неспецифического связывания необходимо вычитать из значений полученных радиоактивностей для всех калибровочных проб.
Неспецифическое связывание определяют следующим образом: в пробирки (от 8 до 10 шт.) вносят нулевую калибровочную пробу (или любую калибровочную пробу, или сыворотку крови человека) и все компоненты изделия, предусмотренные инструкцией по его применению, за исключением антисыворотки (антител). Затем проводят все процедуры, предусмотренные инструкцией по применению изделия.
Значение неспецифического связывания , %, рассчитывают по формуле:
![]() , (4)
, (4)
где - среднее арифметическое значение скорости счета в отдельных пробирках, имп/мин;
- среднее арифметическое значение тотальной радиоактивности, имп/мин.
Значение неспецифического связывания должно быть не более 5%.
При использовании твердофазного метода значение неспецифического связывания не определяют.
5.4.1.5 Соотношение скоростей счета калибровочных проб
Для оценки правильности работы системы для радиоиммунологического анализа необходимо проводить определение соотношения скоростей счета (имп/мин) калибровочных проб. Значения скоростей счета калибровочных проб могут иметь прямую зависимость от концентрации анализируемого антигена (или антител): ![]() (сэндвич-вариант), или обратную:
(сэндвич-вариант), или обратную: ![]() (метод конкурентного связывания).
(метод конкурентного связывания).
5.4.1.6 Соотношение скоростей счета калибровочных проб с максимальным и минимальным содержанием антигена (антител) и калибровочной пробы, не содержащей антиген (антитела)
Наклон калибровочной кривой в данной точке является одним из факторов, который определяет разрешающую способность и точность системы для измерения соответствующей концентрации анализируемого антигена. Значение соотношения скоростей счета калибровочной пробы с минимальным содержанием антигена и калибровочной пробы, не содержащей антиген ![]() , может влиять на значение чувствительности системы для радиоиммунологического анализа. Соотношение же скоростей счета калибровочной пробы с максимальным содержанием антигена и калибровочной пробы, не содержащей антиген
, может влиять на значение чувствительности системы для радиоиммунологического анализа. Соотношение же скоростей счета калибровочной пробы с максимальным содержанием антигена и калибровочной пробы, не содержащей антиген ![]() , хотя и не отражает разрешающую способность отдельных участков калибровочного графика, однако является одним из показателей стабильности системы для радиоиммунологического анализа при сравнении разных изделий одной серии и изделий разных серий.
, хотя и не отражает разрешающую способность отдельных участков калибровочного графика, однако является одним из показателей стабильности системы для радиоиммунологического анализа при сравнении разных изделий одной серии и изделий разных серий.
Допускается определять соотношение скоростей счета калибровочной пробы с минимальным содержанием антигена и калибровочной пробы с максимальным содержанием антигена ![]() и соотношение скоростей счета предпоследней калибровочной пробы (
и соотношение скоростей счета предпоследней калибровочной пробы () и калибровочной пробы с максимальным содержанием антигена
![]() .
.
5.4.1.7 Чувствительность
Значение чувствительности определяют следующим образом: в пробирки (от 8 до 10 шт.) вносят нулевую калибровочную пробу и все компоненты изделия, предусмотренные инструкцией по его применению, после чего проводят все процедуры определения. На основании полученных данных вычисляют среднее квадратическое отклонение по формуле:
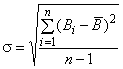 , (5)
, (5)
где - значение скорости счета меченого соединения каждого измерения в пробирках с нулевой калибровочной пробой, имп/мин;
- среднее арифметическое значение скорости счета меченого соединения в пробирках с нулевой калибровочной пробой, имп/мин;
- знак суммирования;
- число определений.
На оси ординат калибровочного графика откладывают значение ![]() - если применяют принцип конкурентного связывания, или
- если применяют принцип конкурентного связывания, или ![]() - если используют сэндвич-вариант; из полученной точки проводят прямую, параллельную оси абсцисс, до пересечения с калибровочным графиком; из точки пересечения опускают перпендикуляр на ось абсцисс.
- если используют сэндвич-вариант; из полученной точки проводят прямую, параллельную оси абсцисс, до пересечения с калибровочным графиком; из точки пересечения опускают перпендикуляр на ось абсцисс.
Соответствующая этому значению концентрация характеризует чувствительность, которую следует выражать в значениях концентрации анализируемого вещества.
Значение чувствительности зависит от крутизны калибровочного графика в области низких концентраций, от точности определения нулевой калибровочной пробы, от константы ассоциации специфических антител (чем больше константа ассоциации, тем чувствительнее система для радиоиммунологического анализа), а также от значения соотношения скоростей счета калибровочной пробы с минимальным содержанием определяемого антигена и нулевой калибровочной пробы.
5.4.1.8 Тест на "открытие" (метод добавок)
При постановке теста на "открытие" смешивают, как правило, равные объемы контрольной сыворотки (плазмы) и калибровочной пробы. Вместо контрольной сыворотки (плазмы) можно использовать калибровочную пробу с минимальной концентрацией определяемого вещества.
Тест на "открытие" , %, проводят в восьми-десяти повторностях. В исследуемой пробе определяют концентрацию анализируемого антигена и сравнивают ее значение с расчетным по формуле:
![]() , (6)
, (6)
где - полученное по калибровочному графику (практическое) значение концентрации анализируемого антигена в исследуемой пробе;
- расчетное (теоретическое) значение концентрации антигена в исследуемой пробе.
Значение "открытия" должно находиться в пределах от 90% до 110%.
Тест на "открытие" является одним из основных для решения вопроса о специфичности и правильности проведения анализа. Важность данного теста заключается также и в том, что при его постановке сравнивают, как взаимодействуют анализируемый антиген, находящийся в исследуемой пробе (например, в контрольной сыворотке или плазме), и анализируемый антиген, находящийся в калибровочной пробе, с антителом. Если они взаимодействуют по-разному, что называется "матриксным эффектом", то появляются систематические ошибки и непостоянство результатов определения.
В случае экспериментальной проверки изделий, предназначенных для определения антигена (антител) в сухих пятнах крови, тест на "открытие" не проводят.
5.4.1.9 Тест на "линейность"
Этот тест заключается в последовательных разведениях калибровочной пробы с максимальной концентрацией антигена или контрольной сыворотки (плазмы) с высокой концентрацией этого антигена в 2, 4, 8 и т.д. раз (возможны и другие пропорции разведения) с последующим сравнением полученных результатов с ожидаемыми при этих разведениях концентрациями анализируемого антигена. Разведения должны быть выполнены с таким расчетом, чтобы тест охватывал все участки калибровочного графика, поэтому наряду с максимальной калибровочной пробой можно использовать калибровочные пробы со средней и даже низкой концентрацией анализируемого вещества. Для разведения следует использовать нулевую калибровочную пробу или сыворотку (плазму) крови, не содержащую исследуемый антиген.
Тест на "линейность" %, проводят в восьми-десяти повторностях. В анализируемых образцах определяют концентрацию исследуемого антигена, умножают на коэффициент разведения и сравнивают ее значение с расчетным по формуле:
![]() , (7)
, (7)
где - полученное по калибровочному графику (практическое) значение концентрации анализируемого антигена в исследуемой пробе;
- расчетное (теоретическое) значение концентрации антигена в исследуемой пробе.
Значение "линейности" должно находиться в пределах от 90% до 110%.
Тест на "линейность" важен тем, что позволяет проконтролировать, насколько правильно калиброваны калибровочные пробы относительно друг друга. При разведении одной калибровочной пробы идет сравнение этой пробы с калибровочными пробами, близкими по концентрации к полученным результатам.
Тест на "линейность" позволяет устранить систематические ошибки, связанные с различной специфичностью анализа, на каждой стадии калибровки, а также позволяет оценить правильность работы системы для радиоиммунологического анализа на всех участках калибровочного графика.
В случае экспериментальной проверки качества изделия, предназначенного для определения антигена (антител) в сухих пятнах крови, тест на "линейность" не проводят.
5.4.1.10 Коэффициент вариации
При определении коэффициента вариации следует использовать аттестованные контрольные сыворотки (плазмы, сухие пятна крови) с низким, средним или высоким содержанием анализируемого антигена. Контроль воспроизводимости проводят в восьми-десяти повторностях. Воспроизводимость проверяют как для одного изделия, так и для разных изделий одной серии и изделий разных серий.
Значение коэффициента вариации , %, определяют по формуле:
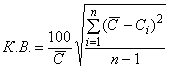 , (8)
, (8)
где - значение каждого измерения концентрации анализируемого антигена в контрольной сыворотке;
- среднее арифметическое значение концентрации анализируемого антигена в контрольной сыворотке;
- знак суммирования;
- число определений.
Воспроизводимость результатов считается удовлетворительной, если значение не превышает 8% при проведении анализа в биологических жидкостях и 15% - при проведении анализа в сухих пятнах биологических жидкостей.
5.4.1.11 Интерсепт
Значение интерсепта определяют по калибровочному графику, построенному в координатах logit-log (на оси ординат откладывают значение соотношения скоростей счета калибровочных проб - и нулевой калибровочной пробы -
, а на оси абсцисс - значение концентрации антигена в логарифмическом масштабе). Если работа изделия основана на принципе конкурентного связывания, то используют соотношение
![]() , если на принципе сэндвич-варианта - соотношение
, если на принципе сэндвич-варианта - соотношение ![]() . Значение интерсепта выражают в тех же единицах, что и значение концентрации анализируемого антигена.
. Значение интерсепта выражают в тех же единицах, что и значение концентрации анализируемого антигена.
При контроле качества изделий для радиоиммунологического анализа обычно пользуются тремя значениями интерсепта - 20% (25%), 50% и 80% (75%), хотя допускаются и другие варианты. Отрезок калибровочного графика, ограниченный значениями 20% и 80% интерсепта, - это "рабочий" отрезок калибровочного графика, являющийся наиболее чувствительной его частью. Значение 50% интерсепта должно лежать на самой крутой части калибровочного графика и должно соответствовать одной и той же средней концентрации анализируемого антигена в пределах установленных колебаний.
Значения интерсепта отражают стабильность концентрации антигена в анализируемой пробе в течение длительного времени; эти значения не должны в большой степени варьировать от изделия к изделию, так как от этого зависит достоверность получаемых в клинической практике результатов при использовании изделий разных серий.
5.4.1.12 Концентрация анализируемого антигена в контрольной сыворотке (плазме, сухом пятне крови)
Для проверки правильности (достоверности) определения концентрации анализируемого антигена в контрольной сыворотке (плазме, сухом пятне крови) используют метод, основанный на применении контрольных сывороток (плазм, сухих пятен крови) с известной концентрацией анализируемого антигена, которые по составу и остальным свойствам должны быть наиболее близки к образцам анализируемого материала (во избежание "матриксного эффекта"). Изделия для радиоиммунологического анализа, как правило, комплектуют контрольными сыворотками с известным содержанием анализируемого антигена. Сравнение полученной в результате определения и указанной в паспорте изделия концентрации позволяет оценить достоверность (правильность) ее определения конкретным изделием для радиоиммунологического анализа.
5.4.2 Изделие для иммунорадиометрического анализа
Экспериментальная оценка изделий включает в себя определение следующих показателей: общая радиоактивность меченого компонента - по формуле (1) или по формуле (2); тотальная (общая) радиоактивность; соотношение скоростей счета калибровочных проб: ![]() ; скорость счета в калибровочной пробе, не содержащей антиген (антитела); отношение скорости счета в калибровочной пробе с максимальным содержанием антигена к тотальной (общей) радиоактивности; "открытие" - по формуле (6); "линейность" - по формуле (7); коэффициент вариации - по формуле (8); интерсепт; концентрация анализируемого антигена (антител) в контрольной сыворотке (плазме). При определении чувствительности рассчитывают значение среднего квадратического отклонения (
; скорость счета в калибровочной пробе, не содержащей антиген (антитела); отношение скорости счета в калибровочной пробе с максимальным содержанием антигена к тотальной (общей) радиоактивности; "открытие" - по формуле (6); "линейность" - по формуле (7); коэффициент вариации - по формуле (8); интерсепт; концентрация анализируемого антигена (антител) в контрольной сыворотке (плазме). При определении чувствительности рассчитывают значение среднего квадратического отклонения () - по формуле (5), после чего на оси ординат калибровочного графика откладывают значение (
![]() ) и из этой точки проводят прямую, параллельную оси абсцисс, до пересечения с калибровочным графиком; из точки пересечения опускают перпендикуляр на ось абсцисс - соответствующая этому значению концентрация характеризует чувствительность.
) и из этой точки проводят прямую, параллельную оси абсцисс, до пересечения с калибровочным графиком; из точки пересечения опускают перпендикуляр на ось абсцисс - соответствующая этому значению концентрация характеризует чувствительность.
5.4.3 Изделия для иммуноферментного анализа
Экспериментальная оценка изделий включает в себя определение следующих показателей: соотношение оптических плотностей () калибровочных проб: (
![]() ) при использовании метода конкурентного связывания, (
) при использовании метода конкурентного связывания, (![]() - при сэндвич-варианте); соотношение
- при сэндвич-варианте); соотношение калибровочной пробы с минимальным содержанием антигена и нулевой калибровочной пробы (
![]() ); соотношение
); соотношение калибровочной пробы с максимальным содержанием антигена и нулевой калибровочной пробы (
![]() ); значение
); значение калибровочной пробе с максимальным значением
; чувствительность - определяют значение
по формуле (5), после чего остальные расчеты ведут так, как указано в 5.4.1; "открытие" - по формуле (6); "линейность" - по формуле (7); коэффициент вариации - по формуле (8); интерсепт; концентрация анализируемого антигена в контрольной сыворотке (плазме). Допускается определять соотношение
калибровочной пробы с минимальным содержанием антигена и калибровочной пробы с максимальным содержанием антигена (
![]() ) и соотношение
) и соотношение предпоследней калибровочной пробы и калибровочной пробы с максимальным содержанием антигена (
![]() ).
).
5.4.4 Изделия для иммунофлюоресцентного анализа
Экспериментальная оценка изделий включает в себя определение следующих показателей: соотношение интенсивностей флюоресценции () калибровочных проб: (
![]() ) при использовании метода конкурентного связывания, (
) при использовании метода конкурентного связывания, (![]() ) - при сэндвич-варианте; соотношение
) - при сэндвич-варианте; соотношение калибровочной пробы с минимальным содержанием антигена и нулевой калибровочной пробы (
![]() ); соотношение
); соотношение калибровочной пробы с максимальным содержанием антигена и нулевой калибровочной пробы (
![]() ); значение
); значение в калибровочной пробе с максимальным значением антигена; чувствительность - определяют значение
по формуле (5), после чего остальные расчеты ведут так, как указано в 5.4.2; "открытие" - по формуле (6); "линейность" - по формуле (7); коэффициент вариации - по формуле (8); интерсепт; концентрация анализируемого антигена в контрольной сыворотке (плазме, сухом пятне крови). Допускается определять соотношение
калибровочной пробы с минимальным содержанием антигена и калибровочной пробы с максимальным содержанием антигена (
![]() ) и соотношение
) и соотношение предпоследней калибровочной пробы (
![]() ) и калибровочной пробы с максимальным содержанием антигена (
) и калибровочной пробы с максимальным содержанием антигена (![]() ).
).
5.4.5 Изделия для иммунохемилюминесцентного анализа
Экспериментальная оценка изделий включает в себя определение тех же показателей, которые используют для проверки изделий для иммуноферментного анализа (см. 5.4.3); при этом определяют значение аналитического сигнала калибровочных и других исследуемых проб.
5.4.6 Изделия для других видов иммунохимического анализа
Экспериментальная оценка изделий (основанных на использовании методов иммунотурбидиметрии, иммунодиффузии и др.) включает в себя определение тех же показателей, которые используют для проверки качества изделий для иммуноферментного анализа (см. 5.4.3).
Описанные выше методы экспериментальной проверки качества изделий для различных вариантов иммунохимического анализа относятся к изделиям для выполнения количественных анализов (исследований).
При проверке качества изделий для качественного или полуколичественного определения особое значение имеет расчет соотношения значений оптических плотностей (или скоростей счета, или интенсивности флюоресценции, или аналитического сигнала) положительного и отрицательного контрольных образцов, входящих в состав изделия. При этом используют следующие варианты:
а) разница значений оптических плотностей отрицательного и положительного контрольных образцов должна быть не более какого-либо заданного значения или же должна находиться в каких-либо заданных пределах;
б) различие в значениях соотношения оптических плотностей отрицательного и положительного контрольных образцов должно быть не менее какого-либо заданного значения или же должно находиться в каких-либо заданных пределах;
в) различие в значениях оптических плотностей отрицательного и положительного контрольных образцов должно быть статистически достоверным (метод контроля основан на вычислении среднего арифметического значения плюс 3).
Поскольку при качественном или полуколичественном иммунохимическом анализе часто используют визуальную оценку результатов, то, наряду с указанными выше методами оценки изделий, основным критерием при выборе метода контроля должно быть заметное различие в интенсивности окраски положительного и отрицательного контрольных образцов.
Экспериментальная оценка изделий, использующих латекс-агглютинацию, если они предназначены для качественного определения анализируемого вещества, включает в себя определение чувствительности, образования агглютинации с латексным реагентом, положительного контроля и отрицательного контроля. В случае полуколичественного определения анализируемого вещества экспериментальная проверка качества этих изделий, наряду с определением чувствительности, образования агглютинации с латексным реагентом, положительного контроля и отрицательного контроля, включает в себя также определение титра анализируемого вещества.
5.4.7 Изделия для иммунохроматографического анализа
Изделия для иммунохроматографического анализа, как правило, предназначены для качественного определения анализируемого вещества.
Экспериментальная оценка изделий включает в себя определение следующих показателей: чувствительность, положительный контроль, отрицательный контроль и время достижения устойчивых показателей теста. При проверке качества этих изделий обязательным является использование международных стандартных образцов, национальных стандартных образцов по ГОСТ 8.315 или образцов исследуемого биоматериала с точно установленным содержанием анализируемого вещества и гарантированно документированных.
5.4.8 Изделия для микроанализа нуклеотидных последовательностей
Экспериментальная оценка изделий включает в себя определение следующих показателей: положительный контрольный образец ДНК (или РНК) и отрицательный контрольный образец. Допускается также использовать маркеры молекулярного веса ДНК (или РНК) и внутренний контроль.
5.4.9 Изделия для фотометрического анализа
5.4.9.1 Растворимость компонентов
В том случае, когда компоненты (реагенты, реактивы), входящие в состав изделия, поставляются в сухом состоянии, необходимо проверять их растворимость: в дистиллированной (бидистиллированной) воде или другом компоненте (реагенте, реактиве) изделия, поставляемом в жидком виде, отметив время растворения.
5.4.9.2 Чувствительность
Для определения чувствительности используют контрольные лабораторные растворы.
Значение чувствительности определяют следующим образом. В пробирки (от 8 до 10 шт.) вносят контрольный лабораторный раствор анализируемого вещества (сыворотку или плазму крови с известной активностью фермента) и все компоненты изделия, предусмотренные инструкцией по его применению, после чего проводят все процедуры определения. Одновременно проводят все процедуры определения калибратора, входящего в состав изделия. После этого измеряют оптическую плотность в каждой пробирке с контрольным лабораторным раствором и с калибратором против холостой пробы (раствор сравнения, состоящий, как правило, из дистиллированной воды или смеси компонентов (реагентов), входящих в изделие, за исключением калибратора).
Среднее арифметическое значение оптической плотности , единицы оптической плотности (ед. опт. плотн.), контрольного лабораторного раствора и калибратора определяют по формуле:
![]() , (9)
, (9)
где - значение
каждого измерения, ед. опт. плотн.;
- знак суммирования;
- число определений.
Концентрацию анализируемого вещества (активность фермента) в контрольном лабораторном растворе определяют по формуле
![]() , (10)
, (10)
где - среднее арифметическое значение
контрольного лабораторного раствора, ед. опт. плотн.;
- среднее арифметическое значение
калибратора, ед. опт. плотн.;
- концентрация анализируемого вещества (активность фермента) в калибраторе.
В ряде случаев, если это предусмотрено инструкцией по применению изделия, допускается определять концентрацию анализируемого вещества (активность фермента) по калибровочному графику.
Для определения активности фермента кинетическим методом по фактору в измерительную кювету спектрофотометра вносят сыворотку (плазму) крови и все компоненты изделия, предусмотренные инструкцией по его применению, инкубируют (в случае необходимости) в течение указанного в инструкции времени. После этого определяют (согласно инструкции) изменение анализируемой пробы во времени.
Изменение (
![]() ) определяют по формуле:
) определяют по формуле:
![]() , (11)
, (11)
где - изменение
пробы, ед. опт. плотн.;
- интервал времени между определениями, мин.
Определение чувствительности в одной и той же пробе сыворотки (плазмы) крови проводят 5 раз.
Среднее арифметическое значение изменения анализируемой пробы рассчитывают по формуле (9).
Активность фермента () определяют по формуле:
![]() , (12)
, (12)
где ![]() - изменение
- изменение пробы, ед. опт. плотн.;
- фактор пересчета,
/дм
.
Фактор пересчета () определяют по формуле:
![]() , (13)
, (13)
где - конечный объем измеряемой пробы, см
;
- объем добавленной пробы сыворотки крови, см
;
- коэффициент молярной экстинкции NADH в условиях определения, см
ммоль
;
10 - коэффициент пересчета для выражения активности фермента в
/дм
; и указывают в паспорте на изделие.
Полученное значение характеризует чувствительность, которую следует выражать в значениях концентрации анализируемого вещества (активности фермента).
Экспериментально установлено, что обычно значение чувствительности в изделиях для фотометрического анализа равно среднему арифметическому значению холостой пробы плюс 3.
5.4.9.3 Соответствие калибратора контрольному лабораторному раствору
В том случае, когда в комплект изделия включен калибратор, не являющийся отдельным коммерческим продуктом, для проверки калибратора необходимо проведение теста на соответствие калибратора контрольному лабораторному раствору анализируемого вещества с концентрацией, равной концентрации калибратора.
В один ряд пробирок (от 5 до 7 шт.) вносят контрольный лабораторный раствор анализируемого вещества, в другой ряд пробирок (от 5 до 7 шт.) - калибратор, после чего добавляют все компоненты изделия, предусмотренные инструкцией по его применению, и проводят все необходимые процедуры определения в соответствии с инструкцией. После этого измеряют значение в каждой пробирке с контрольным лабораторным раствором и с калибратором против холостой пробы. Среднее арифметическое значение
контрольного лабораторного раствора и калибратора определяют по формуле (9). Концентрацию анализируемого вещества определяют по формуле (10). Относительное расхождение между концентрацией анализируемого вещества в контрольном лабораторном растворе
и в калибраторе
, %, определяют по формуле
![]() . (14)
. (14)
Расхождение не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.4 Тест на "открытие"
При постановке теста на "открытие" смешивают, как правило, равные объемы контрольной сыворотки (плазмы) и контрольного лабораторного раствора анализируемого вещества с известной концентрацией, или же равные объемы контрольных лабораторных растворов определяемого вещества с известными концентрациями. В случае определения активности ферментов, как правило, смешивают равные объемы сывороток (плазм) крови человека с известными активностями ферментов.
Тест на "открытие" проводят в восьми-десяти повторностях. В анализируемой пробе определяют концентрацию исследуемого вещества (активность фермента) в соответствии с инструкцией по применению изделия и сравнивают ее значение с расчетным по формуле (6).
Расхождение не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.5 Тест на "линейность"
При проведении этого теста определяют анализируемое вещество (активность фермента) в контрольных лабораторных растворах анализируемого вещества с концентрациями, охватывающими всю область линейного определения, после чего сравнивают полученные значения с теоретическими значениями анализируемого вещества (активности фермента).
Тест на "линейность" проводят в восьми-десяти повторностях по каждому контрольному лабораторному раствору. В анализируемых образцах определяют концентрацию исследуемого вещества (активность фермента) в соответствии с инструкцией по применению изделия и сравнивают ее значения с теоретическими значениями по формуле (7).
Расхождение не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.6 Коэффициент вариации
Для определения коэффициента вариации можно использовать аттестованные контрольные сыворотки (плазмы) с низкой, средней или высокой концентрацией анализируемого вещества (активностью фермента), контрольные лабораторные растворы с известной концентрацией анализируемого вещества (активностью фермента) или образцы биоматериала человека с гарантированно документированной точной концентрацией определяемого вещества (активностью фермента). Определение коэффициента вариации проводят в восьми-десяти повторностях по двум-трем различным концентрациям анализируемого вещества (активностям ферментов). Воспроизводимость проверяют как для одного изделия, так и для изделий одной серии и изделий разных серий. Значение коэффициента вариации определяют по формуле (8).
Коэффициент вариации не должен превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.7 Допустимый разброс результатов при параллельных определениях одной пробы разными изделиями одной серии
Для определения разброса результатов можно использовать аттестованные контрольные сыворотки (плазмы) с низкой, средней или высокой концентрацией анализируемого вещества (активностью фермента), контрольные лабораторные растворы с известной концентрацией анализируемого вещества (активностью ферментов) или образцы биоматериала человека с гарантированно документированной точной концентрацией определяемого вещества (активностью фермента).
Тест проводят в восьми-десяти повторностях по каждому отдельному изделию с использованием реагентов и калибратора соответствующего изделия. Концентрацию анализируемого вещества (активность фермента) определяют по формуле (10). Разброс результатов (), %, при параллельных определениях одной пробы разными изделиями одной серии вычисляют по формулам:
![]() (между изделиями N 1 и 2);
(между изделиями N 1 и 2);
![]() (между изделиями N 1 и 3); (15)
(между изделиями N 1 и 3); (15)
![]() (между изделиями N 2 и 3),
(между изделиями N 2 и 3),
где ,
и
- средние арифметические значения концентрации анализируемого вещества (активности фермента), полученные при использовании изделий N 1, 2 и 3 соответственно.
Допустимый разброс не должен превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.8 Оптическая плотность цветообразующего раствора
Этот тест используют при экспериментальной проверке качества изделий, принцип определения которых основан на образовании окрашенного соединения, подвергаемого фотометрированию.
При выполнении этого теста (в пяти-семи повторностях) измеряют цветообразующего раствора в соответствии с инструкцией по применению изделия. Среднее арифметическое значение
определяют по формуле (9).
Значение не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.9 Время достижения устойчивых значений оптической плотности при проведении цветной реакции
Этот тест используют при экспериментальной проверке качества изделий, принцип определения которых основан на образовании окрашенного соединения, подвергаемого фотометрированию.
При проведении этого теста после завершения всех процедур по определению анализируемого вещества (активности фермента), предусмотренных инструкцией по применению изделия, определяют значение окрашенного продукта реакции, включают секундомер и далее измеряют значения
через равные промежутки в течение заданного времени до момента достижения постоянного значения
. Определение следует повторить не менее трех раз и рассчитать среднее арифметическое значение.
Время достижения устойчивых значений не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.10 Изменение оптической плотности рабочего раствора реагентов
Этот тест используют при экспериментальной проверке качества изделий, принцип определения которых основан на кинетическом измерении уменьшения значения в неокрашенных образцах.
При проведении этого теста, характеризующего скорость автоокисления, измеряют рабочего раствора реагентов при комнатной температуре (от 18 °С до 25 °С) при длине волны и кювете с длиной оптического пути, предусмотренных инструкцией по применению изделия, сразу после приготовления указанного раствора и три раза с интервалом в 30 мин. Определяют среднее арифметическое значение уменьшения
в час при комнатной температуре.
Значение изменения не должно превышать значения, указанного в технической документации на изделия конкретного вида.
5.4.9.11 Межфлаконная вариация
В том случае, если в состав изделия включены компоненты с одинаковым содержанием анализируемого вещества (например, три флакона контрольного образца анализируемого вещества или три флакона контрольной сыворотки), необходимо определить межфлаконную вариацию содержания анализируемого вещества с использованием адекватной методики количественного определения исследуемого вещества (активности фермента). Определение проводят в пяти-семи повторностях в каждом флаконе для каждого анализируемого вещества (активности фермента), после чего рассчитывают средние арифметические значения для каждого флакона по каждому определяемому соединению.
Значение межфлаконной вариации по каждому определяемому соединению не должно превышать значения, указанного в технической документации на изделия конкретного вида.
Описанные выше методы экспериментальной проверки качества изделий для фотометрического анализа относятся к изделиям для выполнения количественных анализов (исследований).
5.4.10 Изделия для качественного или полуколичественного анализа
Экспериментальная оценка изделий для качественного или полуколичественного анализа (это, как правило, индикаторные тест-полоски) используют следующие показатели: чувствительность сенсорного элемента индикаторных полосок; время достижения стабильной окраски сенсорного элемента; правильность определения анализируемого вещества; помехоустойчивость определения анализируемого вещества к наличию других интерферирующих соединений. Для проведения тестов используют контрольные лабораторные растворы с различными известными концентрациями исследуемого вещества, а также контрольные лабораторные растворы, содержащие известные концентрации исследуемого вещества и интерферирующих соединений. Сравнивают окраску сенсорного элемента индикаторных полосок с полями цветовой шкалы, соответствующими определенному содержанию анализируемого вещества.
5.4.11 Изделия для коагулометрического анализа
Изделия для коагулометрического анализа используют для определения различных параметров свертывающей, противосвертывающей и фибринолитической систем.
Экспериментальная оценка изделий включает в себя определение следующих показателей: растворимость компонентов, количественное определение анализируемого параметра, отклонение значений анализируемого параметра от аттестованного значения этого показателя, чувствительность*, "открытие"*, "линейность"*, коэффициент вариации, допустимый разброс результатов при параллельных определениях одной пробы разными изделиями одной серии.
__________________
* За исключением тестов, результаты которых выражены как время свертывания в с.
Для проведения этих тестов используют контрольные плазмы с аттестованным значением в нормальной и патологических областях. Определение проводят в трех-пяти повторностях. Расчетные формулы в основном не отличаются от таковых, применяемых при экспериментальной оценке изделий для фотометрического анализа.
Значения определяемых параметров системы гемостаза не должны превышать по каждому анализируемому параметру значений, указанных в технической документации на изделия конкретного вида.
5.4.12 Микробиологические питательные среды
Оценка соответствия изделия - микробиологическая питательная среда (далее по тексту - среда) проводится с учетом требований ГОСТ Р ЕН 12322 и по методикам, изложенным в Методических указаниях [5]. Безопасность для персонала и окружающей среды при контроле сред необходимо обеспечивать в соответствии с требованиями санитарных правил [6].
Экспериментальная оценка среды включает в себя проведение оценки микробиологических характеристик, исследование функциональных характеристик (специфической активности с помощью контрольных штаммов), а также визуальной оценки (цветность, прозрачность, растворимость), определение значения рН готовой среды, стерильности каждой приготовленной партии (контроль чистоты розлива).
Серия среды признается годной только после проведения всех видов контроля.
5.4.12.1 Растворимость
Количество среды в граммах, необходимое для приготовления конкретной серии питательной среды, должно растворяться при перемешивании и, если необходимо, при кипячении в течение от 2 до 3 минут. Препарат считают растворившимся, если в растворе при визуальном наблюдении в проходящем свете не обнаруживаются частицы вещества.
5.4.12.2 Прозрачность и цветность сухих компонентов изделия
Прозрачность и цветность сухих компонентов среды определяют визуально в проходящем свете в растворе после фильтрации его через ватно-марлевый фильтр (агаровые среды) и бумажный фильтр (жидкости).
5.4.12.3 Прозрачность и цветность
Прозрачность и цветность готовых сред, разлитых во флаконы, определяют визуально в проходящем свете. Агары предварительно расплавляют в кипящей водяной бане и разливают в чашки Петри слоем от 4 до 5 мм.
5.4.12.4 Определение рН
Определение рН питательных сред проводят с помощью потенциометра. Если значение рН изготовленной среды не соответствует требуемым значениям, проводят коррекцию добавлением кислоты или щелочи, или готовят новую партию.
5.4.12.5 Определение стерильности
Определение стерильности готовой среды проводят визуально путем просмотра от 3% до 5% каждой партии после инкубации в термостате при температуре 37 °С в течение от 2 до 14 суток в зависимости от типа среды.
5.4.12.6 Определение чистоты розлива
Среды, приготавливаемые из сухих компонентов и (или) разливаемые в лаборатории в чашки (пробирки), контролируют на чистоту розлива. В зависимости от объёма розлива необходимо проверять от 1 до 2 чашек (пробирок) или 1% чашек или пробирок в начале и столько же в конце процесса розлива среды. Чашки (пробирки) следует инкубировать не менее 18 ч при 37 °С или в условиях инкубирования, рекомендуемых для каждой среды.
5.4.12.7 Определение специфической активности
Контроль специфической активности питательных сред необходимо проводить с применением расширенного набора контрольных штаммов, включающего референтные штаммы. Референтные штаммы могут быть получены из специализированных коллекций:
- Государственная коллекция патогенных микроорганизмов (ФГБУ ГИСК им.Л.А.Тарасевича Роспотребнадзора на базе ФГБУ "Научный центр экспертизы средств медицинского применения" Министерства здравоохранения и социального развития Российской Федерации);
- Всероссийская коллекция промышленных микроорганизмов (ВКПМ) ФГУПГосНИИГенетика;
- Всесоюзная коллекция непатогенных микроорганизмов (ИБФМ);
- Американская коллекция типовых культур (АТСС);
- Английская национальная коллекция типовых культур (NCTC).
Изоляты микроорганизмов, выделенные из клинического материала и используемые для контроля, должны по свойствам соответствовать микроорганизмам признанных национальных и международных коллекций.
Перечень (панель) контрольных штаммов, необходимая для контроля качества каждой питательной среды, определяется ее назначением, т.е. должен состоять, как правило, из нескольких штаммов, моделирующих натурные условия применения среды.
Применяемые для контроля штаммы (из лиофилизированного состояния или со среды хранения) не должны проходить более трёх последовательных пересевов на питательные среды, а свежевыделенные штаммы с момента выделения и получения их в виде чистых культур не должны проходить более 3-5 последовательных пересевов. Они должны быть типичными по культуральным, морфологическим, биохимическим и серологическим тинкториальным свойствам, а также проверяться на отсутствие диссоциации.
Готовую среду засевают культурой (штаммом) целевого микроорганизма (10-10
КОЕ/мл), для которого приготовлена среда и штаммов-ассоциантов (10
-10
КОЕ/мл) и визуально (или под малым увеличением микроскопа) изучают характер роста.
Рост микроорганизмов оценивают с помощью количественных (для плотных сред), полуколичественных или качественных методов. Количество образцов одной серии среды для контроля - не менее трех.
Для всех типов сред (сухие, готовые, плотные, жидкие, дифференциальные, селективные, транспортные, для культивирования и т.д.) определяют чувствительность, скорость роста культуры, а также стабильность основных биологических свойств штамма при росте на исследуемой среде.
5.4.12.8 Скорость роста
Скорость роста (ч) определяют по минимальному времени инкубации посевов, за которое при соответствующем разведении обеспечивается отчетливый (не менее 100 жизнеспособных клеток) видимый невооруженным глазом рост культуры (помутнение, наличие пленки, осадка, роста по уклону и др.) во всех засеянных пробирках с жидкими (полужидкими) питательными средами или формирование типичных, легко дифференцируемых колоний на чашках (пробирках) с плотной средой.
Полуколичественная оценка роста должна быть выполнена с помощью шкалы роста, например от 0 до 3 плюсов (+).
5.4.12.9 Чувствительность среды
Чувствительность среды определяют как максимальное разведение культуры (штамма), обеспечивающее визуально обнаруживаемый рост колоний искомого штамма в течение определенного времени и температуре на всех засеянных чашках (пробирках) с питательной средой.
Качественным методом определяют наличие и характер роста каждого из контрольных штаммов. Рост целевых контрольных штаммов должен быть типичным по цвету, размеру и морфологии колоний; рост нецелевых должен частично или полностью подавляться.
Для оценки результатов количественным методом при интерпретации ингибирующих, накопительных или задерживающих рост свойств питательной среды, подсчитывают количество выросших колоний на тестируемой и контрольной (среде выращивания) средах и определяют ингибирующие, дифференцирующие, нейтрализующие и/или иные соответствующие назначению среды свойства.
5.4.12.10 Дифференцирующие свойства питательной среды
Для сред, предназначенных для идентификации, выделения и дифференциации определяют также дифференцирующие свойства - выраженность отличительных признаков патогенных микроорганизмов от непатогенных видов и естественных ассоциантов (по структурным особенностям колоний, их окраске, изменению цвета и др. признакам).
Дифференцирующие свойства определяют по следующим тестам:
а) выраженность дифференцирующего признака - структура колоний, цвет колоний, изменение цвета среды под колониями, ореол вокруг них, диаметр последнего, появление диффузного изменения цвета среды;
б) четкость дифференциации колоний группы патогенных микроорганизмов от непатогенных, входящих в тот же систематический таксон, и от естественных ассоциантов при посеве смесей.
5.4.12.11 Ингибирующие свойства среды
Ингибирующие свойства определяют для селективных сред как степень подавляющего воздействия на рост и (или) проявление типичных свойств сопутствующей микрофлоры.
Показатель ингибирующих свойств выражают как минимальное разведение культуры, при посеве из которого полностью отсутствует рост и (или) проявление типичных свойств посторонней микрофлоры на испытуемой среде при его наличии на среде выращивания (ингибирующие свойства), или как величину отношения среднего числа сформировавшихся колоний контрольного штамма на "неингибиторной" среде (среда выращивания) к среднему числу колоний на испытуемой "ингибиторной" среде (показатель ингибиции) с учетом разведения.
5.4.12.12 Эффективность среды
Среды, предназначенные для накопления микроорганизмов (накопительные питательные среды), оценивают по их эффективности. Эффективность среды характеризуется показателем эффективности () - выход микробных клеток с 1 мл питательной среды (в млрд/мл) и прирост числа микроорганизмов относительно засеянного.
Оценку качества накопительных питательных сред по показателю эффективности производят путем определения концентрации микробных клеток в среде после соответствующей инкубации.
Прирост числа микроорганизмов в накопительной питательной среде в процессе инкубации посевов в течение соответствующего времени () определяют по формуле (в %):
![]() , (16)
, (16)
где - среднее число колоний на чашках после инкубации культуры в жидкой (полужидкой) накопительной среде;
- среднее число колоний при "нулевом посеве";
- степень разведения.
5.4.12.13 Показатели сохранения жизнеспособности и стабильности основных биологических свойств микроорганизмов
Для транспортных сред и сред для хранения (консервирующих), задерживающих размножение и рост микроорганизмов, определяют показатели сохранения жизнеспособности и стабильности основных биологических свойств микроорганизмов.
Посев материала на среды и последующую идентификацию выделенных культур проводят в соответствии с методиками, изложенными в действующих МУ, инструкциях, приказах по диагностике заболеваний, вызываемых соответствующими микроорганизмами.
Для оценки качества питательной среды возможно использование среды сравнения - среды, ранее отконтролированной и удовлетворяющей требованиям качества.
Показатель прорастания микробных клеток определяют как отношение среднего числа колоний, образовавшихся на испытуемой среде, к среднему числу колоний на контрольной среде, выраженное в процентах.
При этом плотные агаровые среды годны к использованию, если показатель прорастания не менее 70%, а на жидких средах наблюдают визуально одинаковый рост контрольного штамма.
Библиография
[1] | GHTF/SG1/N071:2012* Definition of the Terms 'Medical Device' and 'In Vitro Diagnostic (IVD) Medical Device | |
________________ * Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - . | ||
[2] | Федеральный Закон от 26 июня 2008 года N 102-ФЗ "Об обеспечении единства измерений" | |
[3] | Руководство ИСО/МЭК 99:2007 | Международный словарь по метрологии. Основные и общие понятия и соответствующие термины (VIM) |
[4] | Клиническая лабораторная диагностика: Национальное руководство. В 2 томах. Под редакцией В.В.Долгова, В.В.Меньшикова ГЭТАР-Медиа, 2012 | |
[5] | Методические указания | Методы контроля бактериологических питательных сред |
[6] | Санитарно-эпидемиологические правила СП 1.3.2322-08 (с дополнениями и изменениями N 1 СП 1.3.2518) | Безопасность работы с микроорганизмами III-IV групп патогенности (опасности) и возбудителями паразитарных болезней |
УДК 61:006.354 | ОКС 11.020 |
Ключевые слова: диагностика ин витро, методы испытаний | |
Электронный текст документа
и сверен по:
, 2014

{kind=link}