МЕЖГОСУДАРСТВЕННЫЙ СОВЕТ ПО СТАНДАРТИЗАЦИИ, МЕТРОЛОГИИ И СЕРТИФИКАЦИИ (МГС)
INTERSTATE COUNCIL FOR STANDARDIZATION, METROLOGY AND CERTIFICATION (ISC)
ГОСТ 7619— 2023
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ШПАТ ПЛАВИКОВЫЙ
Методы физико-химического анализа
Издание официальное
Москва Российский институт стандартизации 2023
Предисловие
Цели, основные принципы и общие правила проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0 «Межгосударственная система стандартизации. Основные положения» и ГОСТ 1.2 «Межгосударственная система стандартизации. Стандарты межгосударственные, правила, рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены»
Сведения о стандарте
1 РАЗРАБОТАН Ассоциацией «Объединение производителей, поставщиков и потребителей алюминия» (Алюминиевая Ассоциация) совместно с Ассоциацией «Некоммерческое партнерство Координационно-информационный центр государств — участников СНГ по сближению регуляторных практик (Ассоциация «НП КИЦ СНГ»)
2 ВНЕСЕН Межгосударственным техническим комитетом по стандартизации МТК 99 «Алюминий»
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 31 мая 2023 г. № 162-П)
За принятие проголосовали:
Краткое наименование страны по МК (ИСО 3166) 004—97 | Код страны по МК (ИСО 3166) 004—97 | Сокращенное наименование национального органа по стандартизации |
Армения | АМ | ЗАО «Национальный орган по стандартизации и метрологии» Республики Армения |
Беларусь | BY | Госстандарт Республики Беларусь |
Казахстан | KZ | Госстандарт Республики Казахстан |
Киргизия | KG | Кыргызстандарт |
Россия | RU | Росстандарт |
Узбекистан | UZ | Узстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 29 июня 2023 г. № 450-ст межгосударственный стандарт ГОСТ 7619—2023 введен в действие в качестве национального стандарта Российской Федерации с 1 ноября 2023 г.
5 ВЗАМЕН ГОСТ 7619.0—81, ГОСТ 7619.4—81, ГОСТ 7619.5—81, ГОСТ 7619.9—81, ГОСТ 7619.10—75, и ГОСТ 19724—74
ГОСТ 7619.1—74,
ГОСТ 7619.6—81,
ГОСТ 7619.11—77,
ГОСТ 7619.2—81,
ГОСТ 7619.7—81,
ГОСТ 7619.12—77,
ГОСТ 7619.3—81,
ГОСТ 7619.8—81,
ГОСТ 7619.13—91
Информация о введении в действие (прекращении действия) настоящего стандарта и изменений к нему на территории указанных выше государств публикуется в указателях национальных стандартов, издаваемых в этих государствах, а также в сети Интернет на сайтах соответствующих национальных органов по стандартизации.
В случае пересмотра, изменения или отмены настоящего стандарта соответствующая информация будет опубликована на официальном интернет-сайте Межгосударственного совета по стандартизации, метрологии и сертификации в каталоге «Межгосударственные стандарты»
© Оформление. ФГБУ «Институт стандартизации», 2023

В Российской Федерации настоящий стандарт не может быть полностью или частично воспроизведен, тиражирован и распространен в качестве официального издания без разрешения Федерального агентства по техническому регулированию и метрологии
Содержание
1 Область применения
2 Нормативные ссылки
3 Общие требования к методам химического анализа
4 Методы определения влаги
5 Методы определения карбоната кальция
6 Методы определения фторида кальция
7 Методы определения диоксида кремния
8 Метод определения металлических оксидов Ме2О3
9 Методы определения железа
10 Метод определения серы (общей)
11 Методы определения серы (сульфидной)
12 Методы определения фосфора
13 Методы определения содержания оксида магния
14 Метод определения содержания оксида стронция
15 Метод определения содержания оксида бария
16 Метод определения флотационных реагентов
17 Метод определения гранулометрического состава
МЕЖГОСУДАРСТВЕННЫЙ СТАНДАРТ
ШПАТ ПЛАВИКОВЫЙ
Методы физико-химического анализа
Fluorite. Methods of physicochemical analysis
Дата введения — 2023—11—01
1 Область применения
Настоящий стандарт распространяется на шпат плавиковый и устанавливает общие требования к методам химического анализа и методу определения гранулометрического состава.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие межгосударственные стандарты:
ГОСТ 12.1.004 Система стандартов безопасности труда. Пожарная безопасность. Общие требования
ГОСТ 12.1.005 Система стандартов безопасности труда. Общие санитарно-гигиенические требования к воздуху рабочей зоны
ГОСТ 12.1.007 Система стандартов безопасности труда. Вредные вещества. Классификация и общие требования безопасности
ГОСТ 61 Реактивы. Кислота уксусная. Технические условия
ГОСТ 83 Реактивы. Натрий углекислый. Технические условия
ГОСТ 195 Реактивы. Натрий сернистокислый. Технические условия
ГОСТ 1277 Реактивы. Серебро азотнокислое. Технические условия
ГОСТ 1381 Уротропин технический. Технические условия
ГОСТ 2821 Стронций углекислый. Технические условия
ГОСТ 3118 Реактивы. Кислота соляная. Технические условия
ГОСТ 3158 Реактивы. Барий сернокислый. Технические условия
ГОСТ 3306 Сетки с квадратными ячейками из стальной рифленой проволоки. Технические условия
ГОСТ 3760 Реактивы. Аммиак водный. Технические условия
ГОСТ 3765 Реактивы. Аммоний молибденовокислый. Технические условия
ГОСТ 3776 Реактивы. Хрома (VI) оксид. Технические условия
ГОСТ 4160 Реактивы. Калий бромистый. Технические условия
ГОСТ 4165 Реактивы. Медь (II) сернокислая 5-водная. Технические условия
ГОСТ 4198 Реактивы. Калий фосфорнокислый однозамещенный. Технические условия
ГОСТ 4199 Реактивы. Натрий тетраборнокислый 10-водный. Технические условия
ГОСТ 4204 Реактивы. Кислота серная. Технические условия
ГОСТ 4208 Реактивы. Соль закиси железа и аммония двойная сернокислая (соль Мора). Технические условия
ГОСТ 4212 Реактивы. Методы приготовления растворов для колориметрического и нефелометрического анализа
ГОСТ 4221 Реактивы. Калий углекислый. Технические условия
ГОСТ 4232 Реактивы. Калий йодистый. Технические условия
ГОСТ 4234 Реактивы. Калий хлористый. Технические условия
Издание официальное
ГОСТ 4328 Реактивы. Натрия гидроокись. Технические условия
ГОСТ 4332 Реактивы. Калий углекислый — натрий углекислый. Технические условия
ГОСТ 4461 Реактивы. Кислота азотная. Технические условия
ГОСТ 4463 Реактивы. Натрий фтористый. Технические условия
ГОСТ 4478 Реактивы. Кислота сульфосалициловая 2-водная. Технические условия
ГОСТ 4530 Реактивы. Кальций углекислый. Технические условия
ГОСТ 4919.1 Реактивы и особо чистые вещества. Методы приготовления растворов индикаторов
ГОСТ 6344 Реактивы. Тиомочевина. Технические условия
ГОСТ 6563 Изделия технические из благородных металлов и сплавов. Технические условия
ГОСТ 6613 Сетки проволочные тканые с квадратными ячейками. Технические условия
ГОСТ 6616 Преобразователи термоэлектрические. Общие технические условия
ГОСТ 67091 Вода дистиллированная. Технические условия
ГОСТ 9147 Посуда и оборудование лабораторные фарфоровые. Технические условия
ГОСТ 9656 Реактивы. Кислота борная. Технические условия
ГОСТ 10163 Реактивы. Крахмал растворимый. Технические условия
ГОСТ 10484 Реактивы. Кислота фтористоводородная. Технические условия
ГОСТ 10652 Реактивы. Соль динатриевая этилендиамин-N, N, N', N'- тетрауксусной кислоты 2-водная (трилон Б). Технические условия
ГОСТ 10929 Реактивы. Водорода пероксид. Технические условия
ГОСТ 11293 Желатин. Технические условия
ГОСТ 14180—80 Руды и концентраты цветных металлов. Методы отбора и подготовки проб для химического анализа и определения влаги
ГОСТ 19627 Гидрохинон (парадиоксибензол). Технические условия
ГОСТ 20448 Газы углеводородные сжиженные топливные для коммунально-бытового потребления. Технические условия
ГОСТ 20490 Реактивы. Калий марганцовокислый. Технические условия
ГОСТ 22867 Реактивы. Аммоний азотнокислый. Технические условия
ГОСТ 25336 Посуда и оборудование лабораторные стеклянные. Типы, основные параметры и размеры
ГОСТ 25664 Метол (4-метиламинофенол сульфат). Технические условия
ГОСТ 27068 Реактивы. Натрий серноватистокислый (натрия тиосульфат) 5-водный. Технические условия
ГОСТ 29220 Концентраты плавиковошпатовые металлургические. Технические условия
Примечание — При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов и классификаторов на официальном интернет-сайте Межгосударственного совета по стандартизации, метрологии и сертификации (www.easc.by) или по указателям национальных стандартов, издаваемым в государствах, указанных в предисловии, или на официальных сайтах соответствующих национальных органов по стандартизации. Если на документ дана недатированная ссылка, то следует использовать документ, действующий на текущий момент, с учетом всех внесенных в него изменений. Если заменен ссылочный документ, на который дана датированная ссылка, то следует использовать указанную версию этого документа. Если после принятия настоящего стандарта в ссылочный документ, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение применяется без учета данного изменения. Если ссылочный документ отменен без замены, то положение, в котором дана ссылка на него, применяется в части, не затрагивающей эту ссылку.
3 Общие требования к методам химического анализа
3.1 Определение содержания компонентов проводят параллельно не менее чем в двух навесках, отобранных от пробы плавикового шпата, подготовленной по ГОСТ 14180 и измельченной до размера частиц, проходящих через сито с сеткой № 0063 по ГОСТ 6613, и высушенной при температуре (105 ± 2) °C до постоянной массы. Одновременно в тех же условиях проводят контрольный опыт для внесения в результат анализа соответствующей поправки.
3.2 Взвешивание навесок и осадков проводят с погрешностью не более 0,0002 г.
3.3 При проведении анализа и приготовлении растворов следует применять реактивы квалификации не ниже ч. д. а. и дистиллированную воду по ГОСТ 6709.
3.4 Титр растворов устанавливают не менее чем по трем навескам исходного вещества.
3.5 Концентрацию растворов приводят в граммах веществ на 1000 см3 раствора (г/дм3).
3.6 В выражении «разбавленная 1:1, 1:2» и т. д. первые цифры означают объемные части кислоты или какого-либо раствора, вторые — объемные части воды.
3.7 Выражение «горячая вода (или раствор)» означает, что жидкость имеет температуру 60 °C — 70 °C, а «теплая вода (или раствор)» — 40 °C — 50 °C.
3.8 Стандартные растворы, применяемые для построения градуировочных графиков, готовят в соответствии с ГОСТ 4212.
3.9 Испытания должны проводиться в условиях лаборатории в соответствии с основными правилами безопасной работы в химической лаборатории.
3.10 Проектирование и устройство освещения в лабораторных помещениях необходимо осуществлять в соответствии с требованиями нормативных документов, действующих на территории государства, принявшего настоящий стандарт (далее — нормативный документ2).
3.11 Пожарная безопасность лабораторных помещений должна обеспечиваться в соответствии с требованиями ГОСТ 12.1.004.
3.12 Все используемые электрические приборы должны соответствовать нормативным документам3. Их эксплуатация — в соответствии с нормативными документами4, и Правилами по охране труда при эксплуатации электроустановок24. Все используемые приборы должны пройти государственную
*5 проверку в соответствии с нормативным документом .
3.13 Работы на атомно-абсорбционном спектрофотометре с использованием пропан-бутанового пламени должны проводиться в соответствии с правилами безопасности в газовом хозяйстве.
3.14 Содержание вредных веществ в воздухе рабочей зоны, образующихся в ходе анализа, не должны превышать предельно допустимых концентраций, указанных в ГОСТ 12.1.005.
3.15 Контроль за содержанием вредных веществ в воздухе рабочей зоны лаборатории необходимо осуществлять в соответствии с ГОСТ 12.1.005 и ГОСТ 12.1.007.
Анализ проб воздуха проводят в соответствии с методами определения вредных веществ в воздухе.
3.16 Обезвреживание и удаление отходов, образующихся в результате проведения анализов, следует осуществлять в соответствии с документацией, утвержденной в установленном порядке.
3.17 Помещения лаборатории, в которых выполняется анализ плавикового шпата, необходимо оборудовать системами вентиляции в соответствии с ГОСТ 12.4.021.
3.18 Работающие с плавиковым шпатом обеспечиваются бытовыми помещениями в соответствии с нормативными документами26.
3.19 К работе в лабораториях допускаются лица не моложе 18 лет, проходящие периодический медицинский осмотр и допущенные по состоянию здоровья к работе с вредными веществами.
3.20 При использовании в качестве реактивов опасных (токсичных, едких и т. п.) веществ следует руководствоваться требованиями безопасности, изложенными в соответствующих нормативных документах и технической документации на указанные материалы.
3.21 Совместно с результатом измерений И/ может быть приведена расширенная неопределенность U измерений: W ± U.
Примечание — Расширенная неопределенность измерений U — параметр, связанный с результатом измерений и характеризующий рассеяние значений, которые можно приписать измеряемой величине. Расширенную неопределенность U рассчитывают по формуле
U = ku, (3.1)
где к — коэффициент охвата, равный 2;
и — стандартная неопределенность измерений, равная величине допустимого расхождения результатов параллельных измерений d.
3.22 Контроль правильности результатов измерений проводят с помощью стандартных образцов, не применяемых для градуировки измерительного оборудования и близких по составу к анализируемой пробе.
Абсолютное значение расхождения между результатом анализа контрольного (стандартного) образца X и принятым опорным (аттестованным) значением Хат не должно превышать значение б:
|Х-Хат|<б. (3.2)
Значение норматива 5 определяют по формуле
5=1,44с/, (3.3)
где d— величина допустимого расхождения результатов параллельных измерений.
Если указанное соотношение не выполняется, эксперимент повторяют. При повторном несоответствии полученных результатов нормативу выполнение анализов прекращают, выясняют причины, приводящие к неудовлетворительным результатам, и устраняют их.
4 Методы определения влаги
4.1 Метод определения влаги А
4.1.1 Сущность методаНастоящий подраздел устанавливает метод определения влаги при массовой доле от 0,70 % до 30,0 %.
4.1.2 Подготовка к проведению испытания
Для определения содержания влаги из пробы плавикового шпата отмеряют две навески массой по 0,1 кг из материала крупностью до 0,315 мм и массой по 1,0 кг из материала крупностью свыше 0,315 до 10 мм.
4.1.3 Аппаратура для проведения испытания
Для проведения анализа применяют:
шкаф сушильный с терморегулятором, обеспечивающий температуру нагрева (105 ± 2) °C;
весы лабораторные с погрешностью не более 30 мг;
чаши выпарительные по ГОСТ 9147 или противни;
эксикатор по ГОСТ 25336.
4.1.4 Проведение анализа
Навеску плавикового шпата помещают ровным слоем толщиной не более двойного размера максимального куска в предварительно высушенную до постоянной массы выпарительную чашу и взвешивают. Чашу с навеской помещают в сушильный шкаф, нагретый до (105 ± 2) °C, и выдерживают в течение 1 ч. Затем чашу с навеской вынимают из сушильного шкафа, охлаждают в эксикаторе до комнатной температуры и взвешивают.
4.1.5 Обработка результатов
4.1.5.1 Массовую долю влаги M/H2q, %, вычисляют по формуле
(т< -т2)-100 ivh2o=LJ--^---’ <4-1)
z т
где т1 — масса чаши с навеской до высушивания, г;
т2 — масса чаши с навеской после высушивания, г;
т — масса навески до высушивания, г.
Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать значений, приведенных в таблице 1.
Таблица 1 — Допустимые расхождения результатов определений
Массовая доля влаги, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,7 до 1,5 включ. | 0,10 | 0,15 |
Св. 1,5 » 5,0 » | 0,3 | 0,4 |
» 5,0 » 10,0 » | 0,4 | 0,5 |
» 10,0 » 15,0 » | 0,5 | 0,6 |
» 15,0 » 30,0 » | 0,6 | 0,7 |
Если расхождения между результатами предел повторяемости превышают приведенную величину, определение повторяют.
За окончательный результат анализа принимают среднее арифметическое результатов двух пределов повторяемости.
4.1.5.2 Границы погрешности определения влаги общей массы двух (параллельных) навесок при уровне доверительной вероятности 95 % для 1,5 % и более 1,5 % равны соответственно ±0,05 % и ±0,1 %.
4.2 Метод определения влаги Б
4.2.1 Назначение методаМетод распространяется на все виды плавикового шпата для производства плавиковой кислоты, керамики, а также три вида плавикового шпата для металлургии — концентраты, брикеты и гравий.
4.2.2 Для данного метода использованы следующие определения:
проба для анализа — проба, приготовленная для определения влаги в соответствии с методом, установленным в ГОСТ 14180.
Примечание — Если определение влаги проводят на всем количестве пробы для испытаний, она может быть также названа навеской;
навеска — представительная часть пробы для испытаний, на которой проводят определение влаги.
4.2.3 Сущность метода
Высушивание навески на воздухе при (105 ± 5) °C до постоянной массы.
4.2.4 Оборудование
Сушильный противень с гладкой и чистой поверхностью, на котором можно разместить пробу массой 1 кг или более толщиной не более 10 мм.
Сушильная печь с терморегулирующим устройством, позволяющим поддерживать температуру в любой точке печи в пределах (105 ± 5) °C. Для обеспечения качественного высушивания печь снабжена вентилятором, обеспечивающим циркуляцию воздуха в печи в такой степени, что общий объем воздуха в ней менялся по крайней мере трижды в течение 1 ч, не вызывая потери пробы.
Взвешивающее устройство с чувствительностью 1 г и более и точностью взвешивания, обеспечивающей требуемую повторяемость результатов.
Взвешивающее устройство должно быть защищено от влияния горячего сушильного противня соответствующим теплоизоляционным материалом.
4.2.5 Пробы для анализа
Пробы для анализа должны отбираться и приготавливаться в соответствии с ГОСТ 14180. Приготавливают две или более пробы на партию руды. Для каждого определения влаги используют все количество каждой пробы для анализа. Масса навески должна быть 1 кг и более, максимальный размер частиц руды — 10 мм.
4.2.6 Проведение анализа
4.2.6.1 Число определений
На каждой навеске проводят одно определение влаги.
4.2.6.2 Определение
Взвешивают сушильный противень с точностью до 1 г.
Размещают навеску на сушильном противне слоем не более 10 мм и немедленно взвешивают с точностью до 1 г. Записывают массу сушильного противня и общую массу противня с навеской.
Рассчитывают и записывают исходную массу навески.
Помещают сушильный противень с навеской в сушильную печь с температурой нагрева (105 ±5) °C и поддерживают эту температуру не менее 2 ч.
Для отжатых осадков выдерживают эту температуру в течение 5 ч. Удаляют сушильный противень с навеской из сушильной печи и сразу же взвешивают в горячем состоянии с целью свести к минимуму реабсорбцию атмосферной влаги. Альтернативно навеска может быть взвешена после охлаждения в сушильном шкафу с плотно прилегающей, обеспечивающей герметичность крышкой. В каждом случае метод взвешивания отмечают. Помещают снова сушильный противень с навеской в сушильную печь. Нагревают в течение 1 ч и повторяют взвешивание. Повторяют эти операции до тех пор, пока разность масс последовательных определений не будет 0,1 % и менее от исходной массы навески.
Примечание — Если серию определений влаги проводят на одном и том же виде плавикового шпата, требуемое время нагревания навески может быть определено по предварительно проведенным контрольным анализам.
4.2.7 Расчет и выражение результатов
4.2.7.1 Определение влаги в каждой навеске
Массовую долю влаги И/н 0, %, вычисляют по следующей формуле и округляют результаты до второго десятичного знака:
IVh2o=^^-100. (4.2)
где т1 — исходная масса навески, г;
т2 — масса навески после высушивания, г.
4.2.7.2 Определение влаги в партии
Массовую долю влаги в партии вычисляют по одному из уравнений, указанных ниже, и округляют результат до первого десятичного знака.
Если определение влаги проводят для каждой подпробы при отборе проб на основе массы, содержание массовой доли влаги в партии И/, %, вычисляют по формуле как среднее арифметическое результатов для всех подпроб с учетом числа точечных проб в каждой подпробе
w=—k—•
(4.3)
/=1
где к — число подпроб;
Nj — число точечных проб в /-й подпробе;
Wj — результат определения массовой доли влаги в /-й подпробе, %.
Если нецелесообразно опробовать всю партию, или целесообразно опробовать партию отдельными частями, неравными по массе, как это имеет место при отборе проб на основе времени, содержание влаги в каждой части определяют независимо и по формуле вычисляют содержание влаги в партии И/, %, как среднее арифметическое отдельных результатов
к
Xmlwl
IV = Ц----
(4-4)
к
/=1
где к — число частей в партии;
rrij — масса /-й частицы;
Wj — результат определения влаги в /-й части, %.
Если определение массовой доли влаги проводят в каждой точечной пробе (навеске), массовую долю влаги в партии IV, %, рассчитывают по формуле как среднее арифметическое результатов, полученных в соответствии с 4.2.7.1, для всех точечных проб
(4.5)
где N — число точечных проб, каждая из которых является представительной для равной пропорции партии;
Wj — результат определения массовой доли влаги в /-й точечной пробе, %.
4.2.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- справочный номер результата;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
5 Методы определения карбоната кальция
5.1 Титриметрический метод А
5.1.1 Сущность методаНастоящий подраздел устанавливает титриметрический метод определения карбоната кальция и других его растворимых в уксусной кислоте соединений (в пересчете на карбонат кальция), при массовой доле его от 0,2 % до 50 % и оксида кальция в обожженных окатышах.
5.1.2 Реактивы, растворы и оборудование для проведения анализа
Для проведения анализа применяют:
кислоту соляную по ГОСТ 3118 и раствор с концентрацией 0,1 моль/дм3;
кислоту уксусную по ГОСТ 61, ледяную, х. ч., разбавленную 1:9;
раствор гидроксида калия с концентрацией 280 г/дм3 или раствор гидроксида натрия с концентрацией 80 г/дм3;
триэтаноламин, разбавленный 1:2;
хлорид калия по ГОСТ 4234;
флуорексон (в виде кислоты);
тимолфталеин или индикатор хром темно-синий;
спирт этиловый (гидролизный) ректификованный;
колбу коническую вместимостью 100 см3;
стакан лабораторный вместимостью от 300 до 500 см3.
5.1.3 Подготовка к проведению испытания
5.1.3.1 Для приготовления индикаторной смеси смешивают и растирают 0,4 г флуорексона, 0,33 г тимолфталеина и 40 г хлорида калия. Если в качестве индикатора применяют хром темно-синий, то для приготовления индикаторной смеси 0,1 г хром темно-синий растирают в фарфоровой ступке с 10 г хлоридом калия.
5.1.3.2 Для приготовления растворов трилон Б с концентрациями 0,025 и 0,05 моль/дм3 в 1000 см3 воды разбавляют 9,3 и 18,6 г сухого вещества соответственно.
5.1.3.3 Титр раствора трилона Б устанавливают по государственному стандартному образцу (ГСО) состава флюоритового концентрата, проведенному из трех навесок через все стадии анализа по разделу 6. Титр 0,025 и 0,05 моль/дм3 растворов трилона Б WT по фториду кальция вычисляют по формуле
(С-К)-т-У2
V-Vv100 ’ где С — аттестованное содержание фторида кальция, %;
К — поправка на растворимость фторида кальция в уксусной кислоте, %;
т — масса навески стандартного образца, г;
V2 — объем аликвотной части испытуемого раствора, см3;
V — объем раствора трилона Б, израсходованный на титрование, см3;
— объем всего испытуемого раствора, см3.
5.1.3.4 Содержание карбоната кальция в реактиве Wc, %, вычисляют по формуле
_ (V-ViVO,0050045-100
----—---------. (5.2)
где V — объем 0,1 н. раствора соляной кислоты, взятый для растворения карбоната кальция, см3;
V1 — объем 0,1 н. раствора гидроксида натрия, израсходованный на титрование избытка соляной кислоты, см3;
0,0050045 — титр 0,1 н. раствора соляной кислоты по карбонату кальция;
т — масса навески карбоната кальция, г.
5.1.4 Проведение анализа
Навеску плавикового шпата массой 0,5 г (или 1 г в окатышах) помещают в коническую колбу вместимостью 100 см3, увлажняют несколькими каплями этилового спирта и приливают 10 см3 разбавленной уксусной кислоты. Колбу накрывают фарфоровой крышкой или часовым стеклом и оставляют на 30 мин на кипящей водяной бане.
Содержимое колбы через каждые 10 мин перемешивают встряхиванием. Затем нерастворимый остаток отфильтровывают на плотный фильтр, содержащий небольшое количество фильтробумажной массы. Фильтрат собирают в высокий стакан вместимостью от 300 до 500 см3. Фильтр с остатком и колбу промывают небольшими порциями холодной воды шесть-семь раз. Общий объем фильтрата и промывных вод должен составить не более 75 см3. Если массовая доля карбоната кальция превышает 15 %, фильтрат из стакана переливают в мерную колбу вместимостью 200 см3. Доливают до метки водой и перемешивают. Отбирают 50 см3 раствора в стакан для титрования и приливают 25 см3 воды. К фильтрату или аликвотной части приливают 1,5 см3 раствора триэтаноламина, 0,01 г индикаторной смеси, 10 см3 раствора гидроксида калия или гидроксида натрия и титруют 0,025 М раствором трилона Б (в окатышах 0,05 М раствором трилона Б) до перехода темно-зеленой окраски в фиолетовую (лучше титровать на черном фоне). При титровании в присутствии индикатора хрома темно-синего титруют до перехода малиновой окраски в сине-фиолетовую.
5.1.5 Обработка результатов
5.1.5.1 Массовую долю карбоната кальция И/СаСОз> %, вычисляют по формуле
w Т-1,2818-V-100 к /£_
^СаСОз =---------(5.3)
где Т — титр 0,025 М раствора трилона Б по фториду кальция;
1,2818 — коэффициент пересчета фторида кальция на карбонат кальция;
V — объем 0,025 М раствора трилона Б, израсходованный на титрование, см3;
т — масса навески плавикового шпата, г;
К — поправка на растворимость фторида кальция в уксусной кислоте в пересчете на карбонат кальция, %.
При содержании карбоната кальция до 1 % К = 0,27, свыше 1 % К = 0,13.
5.1.5.2 Массовую долю оксида кальция WCa0, %, вычисляют по формуле
м/ Г-0,5603-V-100
WCaO-----0.52, (5.4
где Т — титр 0,05 М раствора трилона Б по карбонату кальция;
V — объем 0,05 М раствора трилона Б, израсходованный на титрование, см3;
0,5603 — коэффициент пересчета карбоната кальция на оксид кальция;
m — масса навески обожженных флюоритовых окатышей, г;
0,52 — поправка на растворимость в уксусной кислоте фторосодержащих соединений кальция в пересчете на оксид кальция, %.
5.1.5.3 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблицах 2 и 3.
Таблица 2
Массовая доля карбоната кальция, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,20 до 0,50 включ. | 0,06 | 0,08 |
Св. 0,50 » 1,00 » | 0,08 | 0,10 |
» 1,00 » 3,00 » | 0,15 | 0,20 |
» 3,00 » 10,00 » | 0,25 | 0,30 |
» 10,00 » 20,00 » | 0,3 | 0,4 |
» 20,00 » 50,00 » | 0,5 | 0,6 |
Таблица 3
Массовая доля оксида кальция, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,30 до 0,60 включ. | 0,04 | 0,05 |
Св. 0,60 » 2,00 » | 0,03 | 0,10 |
» 2,00 » 6,00 » | 0,12 | 0,15 |
» 6,00 » 10,00 » | 0,15 | 0,20 |
5.2 Титриметрический метод Б
5.2.1 Назначение методаНастоящий подраздел устанавливает титриметрический метод определения содержания карбонатов в плавиковом шпате для производства плавиковой кислоты.
Метод распространяется на продукты с массовой долей карбонатов, выраженных в виде карбоната кальция, равного или более 0,04 %.
5.2.2 Подготовка пробы для анализа
Для приготовления пробы используют остаток от определения потери массы при 105 °C.
5.2.3 Сущность метода
Обработка пробы раствором соляной кислоты, абсорбция освобожденной диоксида углерода раствором гидроксида бария, нейтрализация избытка щелочи раствором соляной кислоты, добавление точно отмеренного избытка стандартного раствора соляной кислоты для растворения осадка карбоната бария и обратное титрование стандартным раствором гидроксида натрия с использованием в качестве индикатора метилоранжа или смеси метилоранжа с ксиленцианолом.
5.2.4 Реактивы
В процессе анализа используют реактивы только аналитической степени чистоты, дистиллированную воду или воду эквивалентной чистоты без примеси диоксида углерода.
Бутанол.
Азот, без диоксида углерода.
Кислота борная.
Бария хлорид дигидрат раствор ВаС12 • 2Н2О, раствор с плотностью 122 г/дм3.
Кислота соляная с плотностью 1,12 г/см3, раствор с концентрацией примерно 25 % (по массе), приготовленный при разбавлении трех объемов соляной кислоты (с плотностью 1,19 г/см3) двумя объемами воды.
Ртути (II) хлорид, насыщенный раствор.
Калия гидроксид, 20 %-ный раствор (по массе).
Кислота соляная, раствор 36,5 г/дм3.
Натрия гидроксид, раствор 40 г/дм3.
Кислота соляная, стандартный раствор с концентрацией 0,1 моль/дм3.
Натрия гидроксид, стандартный раствор с концентрацией 0,1 моль/дм3.
Метиловый оранжевый, раствор 1 г/дм3.
Раствор смеси метилоранжа и ксилен-цианола.
Раствор 1 г метилоранжа и 1,4 г ксилен-цианола в 500 см3 50 %-ного раствора этанола (по объему).
Раствор фенолфталеин с концентрацией 0,25 г/дм3 в 50 %-ном растворе этанола (по объему).
5.2.5 Оборудование
Аппарат для выделения и поглощения газа (см. рисунок 1), содержащий промывную склянку 4, с кругом из пористого стекла 5 пористостью Р1 или Р2, или аналогичного типа, содержащая раствор гидроксида калия, колбу с тремя горлышками 3, вместимостью 500 см3, снабженную капельной воронкой 1 и обратным холодильником 2, промывные склянки Дрекселя 6.
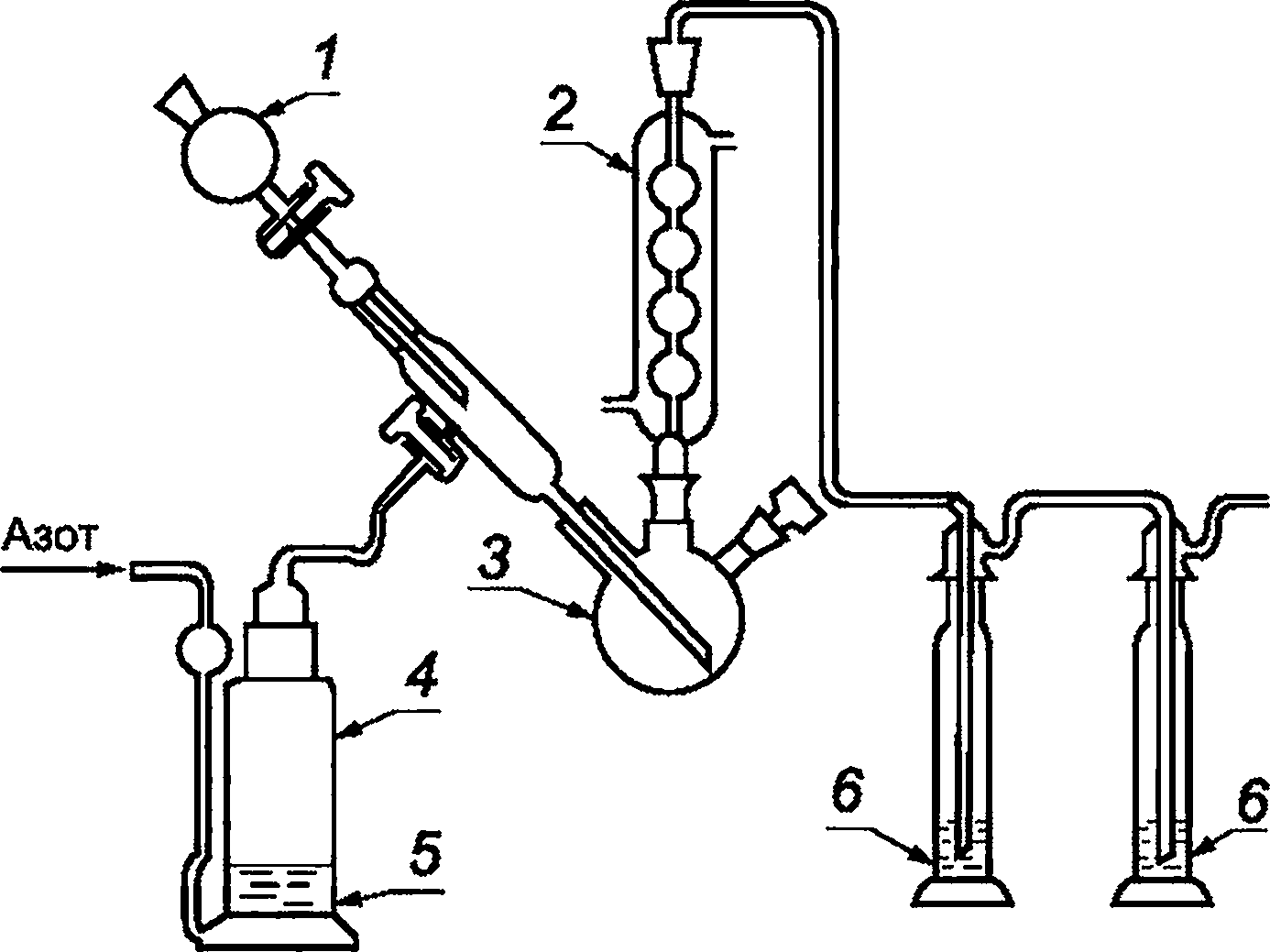
1 — капельная воронка; 2 — обратный холодильник; 3 — колба с тремя горлышками; 4 — промывная склянка;
5 — круг из пористого стекла; 6 — промывная склянка
Рисунок 1 — Пример аппаратов для поглощения газа
Электрошкаф с регулировочным устройством, позволяющим контролировать температуру (105 ± 1) °C.
5.2.6 Проведение анализа
5.2.6.1 Подготовка навески
Измельчают несколько граммов пробы для анализа (5.2.2) в агатовой ступке до размера частиц 63 мкм по ГОСТ 7618. Высушивают просеянный материал в течение 2 ч в электрошкафу при температуре (105 ± 1) °C, охлаждают в эксикаторе и взвешивают примерно 5 г этой пробы до третьего десятичного знака.
Примечание — Важно, чтобы общая масса диоксида углерода, выраженная в виде карбоната кальция, в навеске не превышала 100 мг. Для пробы с содержанием более 2 % масса навески должна быть пропорционально уменьшена.
5.2.6.2 Контрольный опыт
Одновременно с определением проводят контрольный опыт, следуя той же процедуре и используя те же реактивы, но без навески.
5.2.6.3 Определение
Переносят навеску (5.2.6.1) в колбу, используя 100 г воды. Добавляют 4 г борной кислоты и 5 см3 раствора хлорида ртути (II). Закрывают горлышки колбы и пропускают в колбу поток азота со скоростью 50 см3/мин в течение 10 мин.
Не прерывая потока азота, соединяют промывные склянки, в каждой из которых содержится 10 см3 раствора гидроксида натрия, 10 см3 раствора хлорида бария, 1 см3 фенолфталеина, 1 см3 бутанола и 20 см3 воды. Приливают в колбу через капельную воронку 30 см3 соляной кислоты, при необходимости используя резиновый шарик. Перекрывают кран капельной воронки.
Медленно нагревают колбу и слабо кипятят 45 мин. Прекращают нагревание, дают остыть в течение 10 мин, не прерывая поток азота.
Отсоединяют вторую промывную склянку 4 от аппарата, удаляют и ополаскивают входную трубку, собирая в склянку промывные воды. Титруют содержимое склянки раствором соляной кислоты почти до достижения конечной точки.
Примечание — Чтобы избежать абсорбции атмосферного диоксида углерода во время титрования избытка гидроксида натрия в абсорбционном растворе, пропускают поток азота в воздухе над раствором в промывной склянке.
Продолжают титрование раствором соляной кислоты до обесцвечивания фенолфталеина, следя за тем, чтобы не пропустить конечную точку.
Добавляют точно отмеренный объем стандартного раствора соляной кислоты до полного растворения осадка. Погружают входную трубку в этот раствор, чтобы растворить прилипшие частицы карбоната бария, удаляют ее и ополаскивают снова.
Добавляют несколько капель раствора метилоранжа или смеси индикаторов и проводят обратное титрование избытка соляной кислоты стандартным раствором гидроксида натрия.
Нейтрализуют и титруют содержимое первой промывной склянки таким же образом.
5.2.7 Обработка результатов
Массовую долю карбонатов !/ИСаС0 , в процентах по массе карбоната кальция, вычисляют по формуле
_[(Ц-У2)-(У3-У4)]-0.6005
WCaCO3---------------• (5.5)
где У1 — объем стандартного раствора соляной кислоты, использованный для растворения карбоната бария в обеих промывных склянках, см3;
У2 — объем стандартного раствора гидроксида натрия, использованный для обратного титрования избытка соляной кислоты в обеих промывных склянках, см3;
\/3 и V4 — соответственно объемы стандартного раствора соляной кислоты и стандартного раствора гидроксида натрия, использованные для контрольного опыта, см3;
т — масса навесок, г.
Примечание — Если концентрация использованных стандартных растворов не соответствует точно установленным в списке реактивов, следует произвести необходимые поправки.
5.2.8 Подготовка отчета об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
6 Методы определения фторида кальция
6.1 Титриметрический метод
6.1.1 Сущность методаНастоящий подраздел устанавливает титриметрический метод определения фторида кальция при массовой доле свыше 1 %.
Метод основан на разложении смесью борной и соляной кислот остатка, полученного после обработки навески уксусной кислотой при определении карбоната кальция в соответствии с разделом 5. Кальций в фильтрате титруют трилоном Б.
6.1.2 Реактивы и растворы для проведения анализа
Для проведения анализа применяют:
кислоту соляную по ГОСТ 3118, разбавленную 1:3;
кислоту борную по ГОСТ 9656;
калия гидроксид, раствор 280 г/дм3 или раствор гидроксида натрия с концентрацией 80 г/дм3;
соль динатриевую этилендиамин-N, N, N', N'-тетрауксусной кислоты, 2-водную (трилон Б) по ГОСТ 10652, растворы с концентрацией 0,025 и 0,05 М по п.5.3.2;
триэтаноламин, разбавленный 1:2;
хлорид калия по ГОСТ 4234;
флуорексон (в виде кислоты);
тимолфталеин или индикатор хром темно-синий;
смесь индикаторную по 5.1.3.1.
6.1.3 Проведение анализа
6.1.3.1 Остаток, полученный при определении содержания кальция в соответствии с разделом 5, вместе с фильтром переносят в ту же колбу, в которой проводилась обработка уксусной кислотой, прибавляют 1 г борной кислоты, 40 см3 соляной кислоты и при умеренном кипении выдерживают на электроплитке в течение 30 мин (для окатышей 2 г борной кислоты, 50 см3 соляной кислоты — кипятят 45 мин).
6.1.3.2 Горячий раствор фильтруют через фильтр средней плотности в мерную колбу вместимостью 250 см3, промывают горячей водой восемь — десять раз.
6.1.3.3 Раствор охлаждают до комнатной температуры, доливают до метки водой и перемешивают.
6.1.3.4 Отбирают пипеткой 25 см3 раствора в стакан вместимостью 300 — 500 см3, разбавляют водой до 100 см3, приливают 3,5 см3 раствора триэтаноламина, 0,01 г (на кончике шпателя) индикаторной смеси, 10 см3 раствора гидроксида калия или гидроксида натрия и титруют 0,025 М раствором трилона Б (для окатышей — 0,05 М раствором трилона Б) до перехода темно-зеленой окраски раствора в фиолетовую (лучше титровать на черном фоне).
6.1.4 Обработка результатов
6.1.4.1 Массовую долю фторида кальция в плавиковом шпате, за исключением обожженных окатышей H/CaF , %, и фторида кальция в обожженных флюоритовых окатышах , %, вычисляют по формулам:
... УТЦ-100
И/рдр =--1"
VTrVr100
l^CaF =---77-—----+ °-72»
где У — объем 0,025 М раствора трилона Б, израсходованный на титрование, см3;
Т — титр 0,025 М раствора трилона Б, вычисленный по фториду кальция по разделу 5, г/см3;
Tj — титр 0,05 М раствора трилона Б, вычисленный по фториду кальция по разделу 5, г/см3;
V1 — объем всего анализируемого раствора, см3;
У2 — объем аликвотной части анализируемого раствора, см3;
m — масса навески плавикового шпата (за исключением обожженных флюоритовых окатышей), г;
— масса навески обожженных флюоритовых окатышей, г;
К — поправка на растворимость фторида кальция в уксусной кислоте (0,2 — при содержании карбоната кальция до 1 %, 0,1 — свыше 1 %), %;
0,72 — суммарная поправка на растворимость в уксусной кислоте фторида кальция и фторосодержащих соединений в обожженных флюоритовых окатышах, %.
6.1.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятность Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 4.
Таблица 4
Массовая доля фторида кальция, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 1,00 до 3,00 включ. | 0,15 | 0,20 |
Св. 3,00 » 10,00 » | 0,18 | 0,25 |
» 10,00 » 20,00 » | 0,20 | 0,30 |
» 20,00 » 50,00 » | 0,25 | 0,35 |
» 50,00 » 70,00 » | 0,3 | 0,4 |
» 70,00 » 90,00 » | 0,5 | 0,6 |
» 90,00 | 0,6 | 0,7 |
6.2 Потенциометрический метод (после отгонки) определения фтора
6.2.1 Назначение методаНастоящий подраздел устанавливает метод потенциометрического титрования с использованием ион-селективного электрода для определения содержания фтора после его отгонки в плавиковом шпате для производства плавиковой кислоты.
Метод распространяется на материалы с содержанием фторида кальция СаР2 равным или более 90 % (по массе).
6.2.2 Проба для анализа
Для приготовления пробы для анализа используют остаток от определения потери массы при 105 °C.
6.2.3 Сущность метода
Выделение фтора из навески отгонкой в присутствии хлорной кислоты, используя дистилляционный аппарат с регулируемой температурой. Потенциометрическое титрование дистиллята раствором нитрата лантана, с использованием ион-селективного электрода.
6.2.4 Реактивы
В процессе анализа используют реактивы только аналитической чистоты и только дистиллированную воду, или воду эквивалентной чистоты. Для приготовления раствора нитрата лантана используют воду, свободную от диоксида углерода.
Перманганат калия кристаллический.
Натрия фторид перекристаллизованный. Примерно 5 г фторида натрия растворяют в 125 см3 воды и фильтруют раствор под вакуумом посредством малой воронки Бюхнера. Выпаривают раствор в платиновой чашке приблизительно до 60 см3. Охлаждают до 50 °C и отделяют перекристаллизиро-ванный фторид натрия с помощью центрифуги. Промывают кристаллы три раза центрифугированием с небольшим количеством холодной воды. Переносят материал в платиновую чашку и высушивают в электропечи при температуре (105 ± 2) °C. Удаляют чашку из печи, охлаждают в эксикаторе, истирают материал в агатовой ступке и затем просеивают через сито с размером отверстий 355 мкм. Помещают просеянный фторид натрия в платиновую чашку, нагревают в течение 2 ч в электропечи при температуре приблизительно 600 °C и охлаждают в эксикаторе.
Этанол или пропанол.
Кислота хлорная с плотностью 1,54 г/см, 60 %-ный раствор (по массе).
Кислота хлорная, 10 %-ный раствор (по массе). Разбавляют 16,5 см3 раствора хлорной кислоты до 100 см3.
Натрия гидроксид, раствор 1 моль/дм3.
Нитрат лантана, титрованный раствор 0,01 моль/дм3. Растворяют 4,33 г гексагидрата нитрата лантана LaNO3 • 6Н2О в воде. Приливают 10 см3 0,001 моль/дм3 раствора азотной кислоты, количественно переводят в мерную колбу вместимостью 1000 см3, разбавляют водой до метки и перемешивают.
Буферный раствор, pH 6,5. Растворяют 79 г пиридина в 800 см3 воды и нейтрализуют раствором хлорной кислоты до pH (6,5 ± 0,2). Разбавляют водой до 1000 см3 и при необходимости регулируют pH до 6,5.
Фенолфталеин, раствор 5 г/см3 в 95 %-ном растворе этанола (по объему).
6.2.5 Оборудование
Аппарат дистилляционный, включающий парогенератор 3, две электрические нагревательные рубашки 4 и контактный термометр 10 с реле (см. рисунок 2), позволяющий регулировать температуру (135 ± 2) °C в дистилляционной колбе 11. Мощность, необходимая для нагревательных рубашек 4, составляет:
для дистилляционной колбы — 150 Вт;
для парогенератора — минимум 500 Вт.
Целесообразно оборудовать нагревательную рубашку парогенератора регулятором с целью регулирования подводимой мощности.
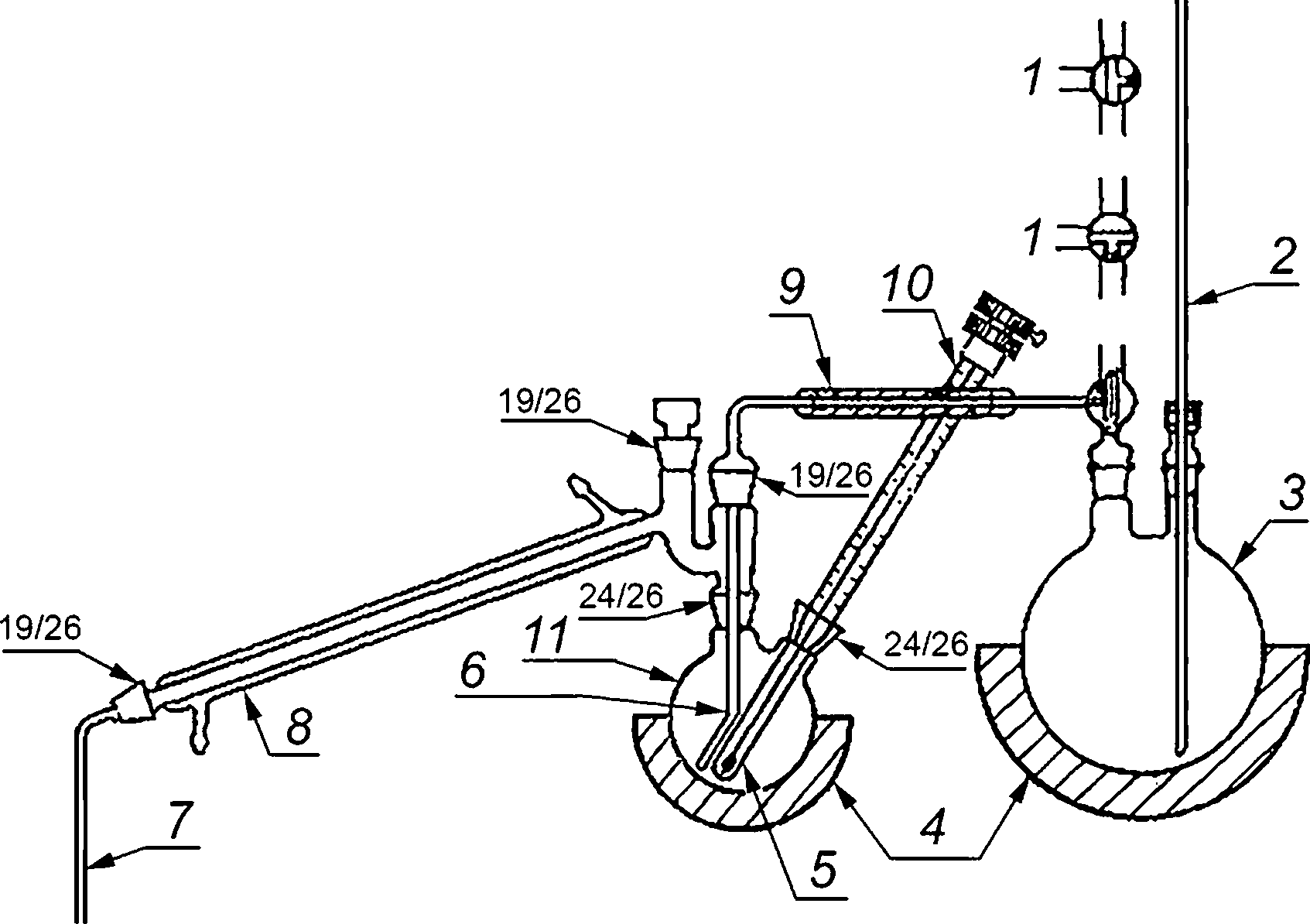
1 — положение кранов 1 и 2; 2— предохранительная трубка; 3— парогенератор; 4 — нагревательная рубашка; 5 — карман для термометра; 6 — пароприемник; 7 — отводная трубка; 8 — конденсатор;
9 — гибкое соединение; 10— контактный термометр; 11 —дистилляционная колба
Рисунок 2 — Дистилляционный аппарат
Мешалка магнитная.
pH-метр, снабженный стеклянным и насыщенным каломельным электродом.
Бюретка вместимостью 20 см3 с ценой деления 0,02 см3.
Электрод фтористый ион-селективный.
Электрод сравнения, насыщенный каломельный или другого типа.
Потенциометр чувствительностью 0,5 мВ, охватывающий диапазон от минус 500 мВ до плюс 500 мВ. При фиксированной конечной точке измерения оборудование с использованием специального набора электродов должно воспроизводить конечную точку в пределах ± 0,5 мВ.
Оборудование автоматическое для регистрации кривой титрования или для титрований до определенного потенциала конечной точки является коммерчески доступным и может быть использовано в качестве альтернативного.
Чашки из боросиликатного стекла с прямыми стенками и плоским дном внутренним диаметром 10 мм, высотой стенок от 10 до 12 мм и толщиной стенок 1 мм.
Печь электрическая, снабженная устройством для регулирования температуры (105 ± 2) °C.
6.2.6 Проведение анализа
6.2.6.1 Навеска
В агатовой ступке истирают несколько граммов пробы по 6.2.2 для анализа до прохождения через сито с размером отверстий 63 мкм. Высушивают просеянный материал в течение 2 ч в печи при температуре (105 ± 2) °C, дают остыть в эксикаторе и взвешивают в одну из чашек примерно 0,2 г с точностью 0,0002 г.
6.2.6.2 Отгонка
Собирают аппарат для дистилляции. Удаляют контактный термометр и пробку из аппарата для перегонки и добавляют несколько кристаллов перманганата калия в дистилляционную колбу. Добавляют 15 см3 воды и 35 см3 хлорной кислоты в дистилляционную колбу и сразу же закрывают аппарат пробкой и вставляют контактный термометр.
Помещают мерную колбу вместимостью 500 см3, содержащую 25 см3 раствора гидроксида натрия и 40 см3 воды, под отводную трубку, которая должна погрузиться в жидкость.
Поворачивают кран между парогенератором 3 и дистилляционным аппаратом в положение крана 1 (верхнее), устанавливают контактный термометр 10 на 135 °C и включают электронагрев дистилляционной колбы 11 и парогенератора 3. Нагревают содержимое дистилляционной колбы до 135 °C в течение приблизительно 15 мин, поворачивают кран в положение 2 (нижнее) и пропускают пар в дистилляционную колбу со скоростью, соответствующей 10 см3 воды в минуту. Собирают примерно 400 см3 дистиллята и после этого прекращают дистилляцию.
Обмывают отводную трубку снаружи и изнутри водой, собирая промывные воды в мерную колбу вместимостью 500 см3. Нейтрализуют дистиллят раствором хлорной кислоты, используя в качестве индикатора несколько капель фенолфталеина. Разбавляют водой до метки и перемешивают.
6.2.6.3 Потенциометрическое титрование
Переводят аликвотную порцию 50,0 см3 из мерной колбы вместимостью 500 см3 в химический стакан вместимостью 250 см3. Добавляют 10 см3 буферного раствора и 60 см3 этанола или пропанола. Вводят магнит магнитной мешалки в химический стакан, помещают стакан на магнитную мешалку, опускают в жидкость фтористый ион-селективный электрод и электрод сравнения (за исключением случая, когда используют комбинированный электрод) и титруют раствором нитрата лантана при норме титрования не более 3,0 дм3/мин, регистрируя объем титранта раствора и соответствующее показание потенциометра. Титруют более медленно в области резко возрастающего изменения потенциала. Оценивают конечную точку графически по вычерченной или зарегистрированной кривой титрования. Альтернативно титруют до определенного потенциала конечной точки, предварительно полученной на основе модели кривых титрования, установленных при идентичных условиях.
Пример типичной кривой титрования приведен на рисунке 3.
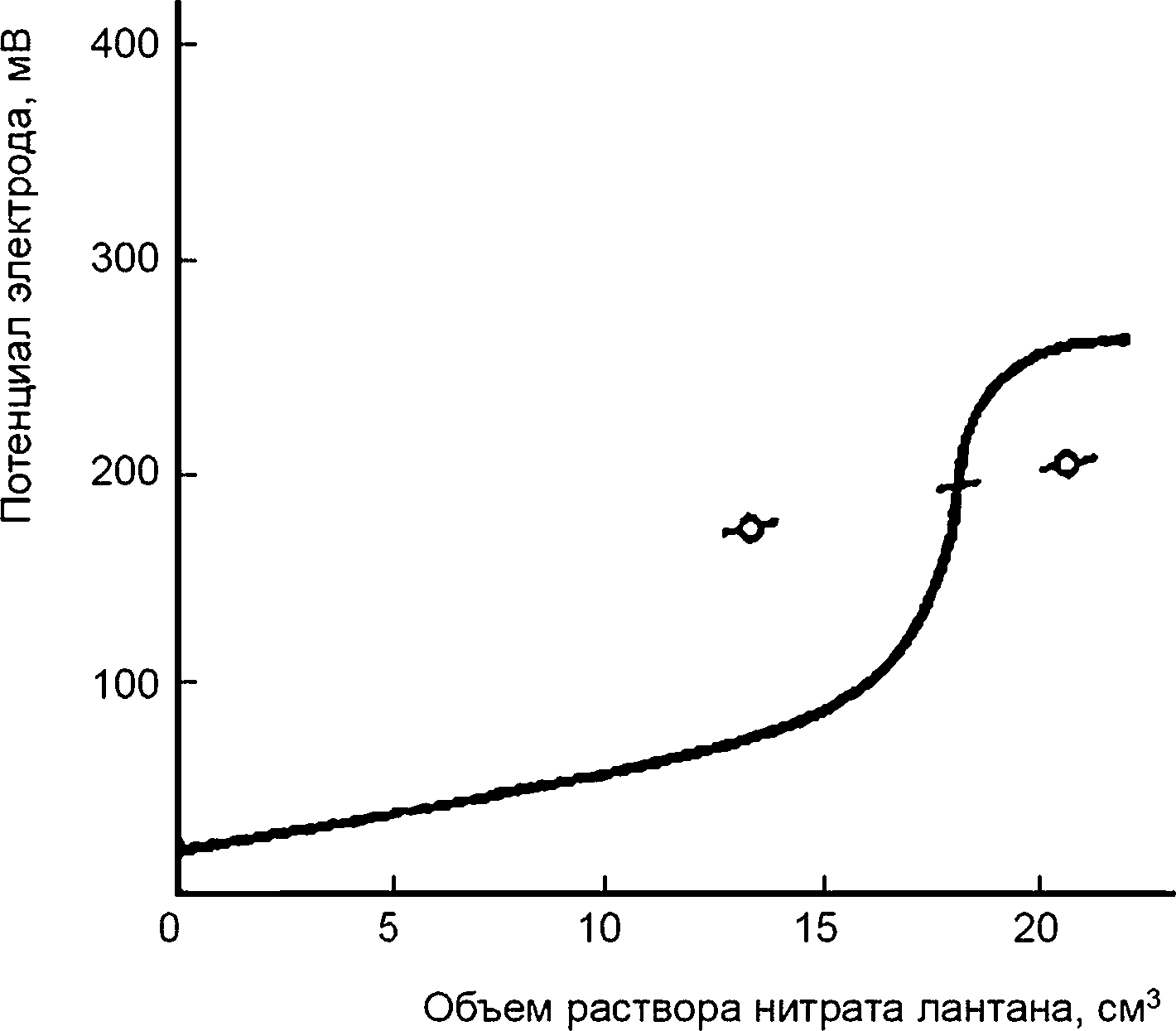
Рисунок 3 —Типичная кривая титрования
6.2.6.4 Установка титра раствора
Выполняют процедуру, описанную в 6.2.6.2, 6.2.6.3, используя вместо навески приблизительно 0,20 г перекристаллизованного фторида натрия, взвешенного с точностью до 0,0001 г. Вычисляют массу фторида натрия, соответствующую 1 см3 раствора азотно-кислого лантана, учитывая результаты контрольного опыта.
6.2.6.5 Контрольный опыт
Одновременно с определением проводят контрольный опыт, используя ту же методику и реактивы, что и при определении, но без навески.
6.2.7 Обработка результатов
6.2.7.1 Вычисления
Массовую долю фтора в процентах фторида кальция И/Сар2 вычисляют по формуле ,„ О,9297 (Ц-Уо)ЮОО
--' <6-3>
а в процентах по массе фтора l/VF — по формуле
О,4524-т,(Ц-Уо)-1ООО_ (64)
/По
где т0 — масса навески, г;
т1 — масса перекристаллизованного фторида натрия, соответствующая 1 см3 раствора нитрата лантана, г;
Уо — объем титрованного раствора нитрата лантана, использованного для контрольного опыта, см3;
— объем титрованного раствора нитрата лантана, использованного для определения, см3.
6.2.7.2 Повторяемость и воспроизводимость
Сравнительные анализы, проведенные в пяти лабораториях на трех пробах, дали статистическую информацию, представленную в таблице 5.
Таблица 5
Наименование показателя | Номер пробы | ||
1 | 2 | 3 | |
Среднее содержание, % Среднее квадратическое отклонение: | 97,04 | 96,79 | 96,41 |
повторяемость | 0,85 | 0,77 | 0,56 |
воспроизводимость | 1,22 | 0,82 | 0,76 |
6.2.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
7 Методы определения диоксида кремния
7.1 Спектрофотометрический метод
7.1.1 Сущность методаНастоящий подраздел устанавливает спектрофотометрический метод определения диоксида кремния при массовой доле от 0,15 % до 50 %.
Метод основан на образовании синего кремнемолибденового комплекса при взаимодействии кремниевой кислоты с молибдатом аммония и восстановлении аскорбиновой кислотой.
7.1.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
печь муфельную, обеспечивающую температуру нагрева (700 ± 25) °C или (1000 ± 50) °C (в зависимости от материала используемых тиглей);
спектрофотометр или фотоэлектроколориметр любого типа для измерения в видимой области спектра;
тигли стеклоуглеродистые вместимостью 50 см3, марки СУ-2500;
тигли железные штампованные или точеные, изготовленные из материала с содержанием кремния менее 0,05 %;
высокие тигли из платины, изделие № 100-7 по ГОСТ 6563;
кислоту соляную по ГОСТ 3118 и разбавленную 1:1, 1:3 и 1:5;
кислоту серную по ГОСТ 4204, растворы 4 моль/дм3 и 0,075 моль/дм3;
кислоту борную по ГОСТ 9656;
карбонат калия, х. ч. или ч. д. а., ГОСТ 4221;
смесь карбонатов калия и натрия, безводный по ГОСТ 4332;
молибдат аммония по ГОСТ 3765, раствор 50 г/см3;
аскорбиновую кислоту, 1 %-ный свежеприготовленный раствор;
желатин пищевой по ГОСТ 11293, 1 %-ный свежеприготовленный раствор;
фенолфталеин по ГОСТ 4919.1, 0,1 %-ный раствор;
натрий кремнекислый мета 9-водный;
натрия перекись;
тетраборат натрия 10-водный, х. ч. или ч. д. а., по ГОСТ 4199, предварительно высушенный при температуре 300 °C до постоянной массы;
карбонат натрия, х. ч. или ч. д. а., по ГОСТ 83;
фторид натрия по ГОСТ 4463;
стандартные растворы диоксида кремния по 7.1.2.1.
7.1.2.1 Приготовление стандартных растворов диоксида кремния
Для приготовления раствора А с содержанием диоксида кремния 1 г/дм3 навеску натрия кремнекислого мета 9-водного массой 4,7301 г растворяют в воде и доливают водой до 1 дм3, перемешивают и переводят в полиэтиленовый сосуд.
Для приготовления раствора Б отбирают пипеткой 20 см3 раствора А в мерную колбу вместимостью 500 см3, доводят до метки водой и перемешивают.
Содержание диоксида кремния в 1 см3 раствора Б (Х^, мг, вычисляют по формуле
7.1.3 Проведение анализа
7.1.3.1 Масса навески плавикового шпата и объем аликвотной части раствора в зависимости от массовой доли диоксида кремния в плавиковом шпате указаны в таблице 6.
Таблица 6
Массовая доля диоксида кремния, % | Масса навески, г | Объем аликвотной части раствора, см3 |
От 0,15 до 1,00 включ. | 1 | 10 |
Св. 1,00 » 2,00 » | 1 | 5 |
» 2,00 » 3,00 » | 0,5 | 10 |
» 3,00 » 5,00 » | 0,5 | 5 |
» 5,00 » 10,00 » | 0,25 | 5 |
» 10,00 » 20,00 » | 0,2 | 5 |
» 20,00 » 30,00 » | 0,2 | 2 |
» 30,00 » 50,00 » | 0,1 | 2 |
7.1.3.2 Навеску плавикового шпата помещают в стеклоуглеродистый тигель, смешивают с 1—2 г борной кислоты, 5 г смеси карбонатов калия и натрия для концентратов и 0,6—0,7 г фторида натрия для остальных продуктов, сплавляют при температуре 700 °C—750 °C в течение 8—10 мин (при сплавлении тигли должны находиться в одинаковой зоне нагрева). Охлажденный плав помещают в стакан вместимостью 400—500 см3. Выщелачивают в 90 см3 соляной кислоты, разбавленной 1:5 (в стакан с плавом приливают 40—45 см3 теплой соляной кислоты, а 45—50 см3 этой кислоты заполняют тигель. Через 2—3 мин содержимое тигля переводят в стакан, обмывая стенки тигля 30—40 см3 воды).
При использовании железного тигля навеску плавикового шпата помещают в тигель, перемешивают с пятикратным количеством перекиси натрия и сплавляют при 650 °C —700 °C в течение 10— 15 мин. Тигель очищают от окалины и теплым помещают в стеклянный стакан вместимостью 400 см3. В стакан осторожно приливают 75—100 см3 теплой воды и закрывают часовым стеклом. После растворения плава тигель ополаскивают водой, раствор немедленно нейтрализуют концентрированной соляной кислотой и добавляют ее в избыток 15 см3. В горячий раствор насыпают 1—2 г борной кислоты.
При использовании платинового тигля навеску плавикового шпата помещают в тигель, присыпают 5 г смеси для сплавления (состоящей из карбоната натрия, безводного тетрабората натрия и карбоната калия в соотношении 1:1:1) и тщательно перемешивают содержимое тигля. Тигель помещают в муфельную печь, предварительно нагретую до 1000 °C. Сплавление проводят в течение 30 мин. Тигель достают из печи, охлаждают и помещают в стакан вместимостью 250 см3. В стакан с тиглем добавляют примерно 100 см3 раствора 1:3 соляной кислоты.
Выщелачивание проводят, часто перемешивая растворы, до полного растворения плава.
Растворы охлаждают и переводят в мерные колбы вместимостью 500 см3, доливают до метки водой и перемешивают. Через 10 мин отбирают аликвотную часть раствора, указанную в таблице 6, и помещают в мерную колбу вместимостью 100 см3. Объем аликвотной части раствора доводят до 15 см3 раствором серной кислоты с концентрацией 0,075 моль/дм3, приливают 5 см3 раствора молибденовокислого аммония, через 10 мин приливают при перемешивании 12—15 см3 4 моль/дм3 раствора серной кислоты, через 2—3 мин 10 см3 раствора аскорбиновой кислоты, доливают до метки водой и перемешивают. Через 10—15 мин измеряют оптическую плотность раствора, применяя светофильтр с максимумом светопропускания 597 нм в кювете с толщиной колориметрируемого слоя 10 мм. Раствором сравнения служит раствор контрольного опыта, проведенный через все стадии анализа. По величине оптической плотности испытуемого раствора находят содержание диоксида кремния по градуировочному графику или методом сравнения со стандартным образцом, близким по составу к анализируемой пробе и проведенным через все стадии анализа.
7.1.3.3 Для построения градуировочного графика в семь мерных колб вместимостью 100 см3 отмеривают бюреткой 1; 2; 4; 6; 8; 10; 12 см3 стандартного раствора Б. В восьмую колбу стандартный раствор не отмеривают.
В каждую колбу приливают по две капли раствора фенолфталеина, нейтрализуют растворы 0,075 моль/дм3 раствором серной кислоты, затем доливают водой до 15 см3, приливают по 0,3 см3 4 моль/дм3 раствора серной кислоты и по 5 см3 раствора молибденовокислого аммония. Через 10 мин приливают при помешивании 30 см3 4 моль/дм3 раствора серной кислоты и далее анализ продолжают, как указано в 7.1.3.2.
Раствором сравнения служит раствор восьмой колбы, который не содержит стандартный раствор диоксида кремния.
По полученным значениям оптической плотности растворов и известным содержаниям диоксида кремния строят градуировочный график.
Правильность построения градуировочного графика проверяют по стандартному образцу флюоритового концентрата.
7.1.4 Обработка результатов
7.1.4.1 Массовую долю диоксида кремния WSiQ2, %, вычисляют по формуле
= .mi:V:100
(7.2)
Sl°2 Vrm-1000
где m1 — количество диоксида кремния, определенное по градуировочному графику, мг;
V — исходный объем испытуемого раствора в мерной колбе, см3;
— объем аликвотной части исходного испытуемого раствора, см3;
т — масса навески плавикового шпата, г.
7.1.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 7.
Таблица 7
Массовая доля диоксида кремния, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,15 до 0,50 включ. | 0,04 | 0,05 |
Св. 0,50 » 1,50 » | 0,08 | 0,10 |
» 1,50 » 3,00 » | 0,12 | 0,15 |
» 3,00 » 5,00 » | 0,15 | 0,20 |
» 5,00 » 10,00 » | 0,20 | 0,25 |
» 10,00 » 30,00 » | 0,3 | 0,4 |
» 30,00 » 50,00 » | 0,4 | 0,6 |
7.2 Спектрометрический кремнемолибденовый метод определения диоксида кремния
7.2.1 Назначение методаНастоящий метод устанавливает спектрометрический кремнемолибденовый метод определения содержания диоксида кремния в плавиковом шпате, используемом для производства плавиковой кислоты и керамики.
Метод применим к продуктам с содержанием кремнезема, выраженным в виде SiO2, от 0,05 % до 4,0 % (по массе).
7.2.2 Проба для анализа
В качестве пробы для испытания должен быть использован остаток, полученный при определении потери массы при температуре 105 °C.
7.2.3 Сущность метода
Разложение навески пробы сплавлением с карбонатом натрия с последующим подкислением соляной кислотой в присутствии борной кислоты для образования фторидного комплекса. Образование молибдокремниевой кислоты и селективное восстановление до синего комплекса молибдокремниевой кислоты с добавлением винной кислоты, чтобы предотвратить мешающее влияние фосфора.
Спектрометрическое измерение окрашенного комплекса при длине волны, соответствующей максимальному поглощению (до 795 нм).
7.2.4 Реактивы
При выполнении анализа необходимо использовать реактивы только аналитической чистоты и дистиллированную воду или воду эквивалентной чистоты. Содержание диоксида кремния в реактивах должно быть очень низким.
Натрия карбонат безводный.
Кислота борная, раствор 40 г/дм3.
Кислота соляная, раствор с концентрацией 7 моль/дм3.
Кислота серная, раствор с концентрацией 7 моль/дм3.
Кислота серная, раствор с концентрацией 18 моль/дм3.
Молибдат, раствор, эквивалентный 55 г молибдена на 1 дм3.
Растворяют 20 г молибденово-кислого аммония [(NH4)6Mo7O24 • 4Н2О] в 150 см3 воды и разбавляют до 200 см3. Раствор сохраняют в колбе и отбрасывают, если появляется осадок.
Кислота винная, раствор 100 г/дм3.
Кислота аскорбиновая, раствор 20 г/дм3. Используют свежеприготовленный раствор.
Силикатный стандартный раствор, соответствующий 500 мг на 1 дм3. В платиновом тигле взвешивают с точностью до 0,0002 г 0,250 г диоксида кремния, полученной при нагревании чистой кремниевой кислоты и прокаленной при температуре 1000 °C до постоянной массы (т. е. до тех пор, пока два последовательных взвешивания не будут отличаться более чем на 0,001 г), или 0,250 г чистого кварца, мелко истертого и предварительно прокаленного в течение 1 ч при температуре 1000 °C и охлажденного в эксикаторе. В тигель добавляют 2,5 г карбоната натрия. Хорошо перемешивают с помощью стеклянной палочки и тщательно сплавляют смесь. Непосредственно в тигель добавляют теплую воду, умеренно нагревают до полного растворения и переливают количественно в химический стакан соответствующей вместимости. Охлаждают, разбавляют раствор примерно до 400 см3, затем переводят в мерную колбу вместимостью 500 см3, добавляют до объема водой и перемешивают. Раствор немедленно переливают в склянку. 1 см3 стандартного раствора содержит 500,0 мкг.
Силикатный стандартный раствор, соответствующий 100 мг на 1 дм3. Помещают 100 см3 стандартного силикатного раствора в мерную колбу вместимостью 500 см3, доводят до объема водой и перемешивают. 1 см3 стандартного раствора содержит 100,0 мкг. Используют свежеприготовленный раствор.
Раствор для разбавления. В химический стакан вместимостью 600 см3 помещают 5 г карбоната натрия и разбавляют примерно в 300 см3 воды. Добавляют 20 см3 раствора борной кислоты и доводят кислотность примерно до pH 2 раствором соляной кислоты с контролем по индикаторной бумаге. Разбавляют водой до 500 см3.
7.2.5 Оборудование
Тигли платиновые диаметром примерно 40 мм и глубиной примерно 30 мм, снабженные платиновыми крышками.
Стаканы химические из материала, не содержащего кремний, вместимостью 100, 250, 600 и 1000 см3.
Склянка из материала, не содержащего кремний.
Колбы мерные из материала, не содержащего кремний.
Стеклянная палочка для перемешивания из материала, не содержащего кремний.
Спектрометр с селектором непрерывного излучения с кюветами с толщиной слоя 20 мм.
Спектрометр с селектором прерывистого излучения с теми же кюветами и фильтрами с областью светопропускания 795 нм. Если нет в наличии таких фильтров, используют фильтр, позволяющий работать при длине волны 680 нм, с кюветами с толщиной слоя 40 мм.
pH-метр со стеклянным измерительным электродом и каломельным электродом, с чувствительностью 0,05 единиц pH.
Электропечь с терморегуляторами, обеспечивающая температуру нагрева до (105 ± 1) °C.
Ступка и пестик из материала, не содержащего кремний, например, оксид алюминия или карбид вольфрама.
7.2.6 Проведение анализа
7.2.6.1 Навеска и приготовление раствора
Истирают несколько граммов пробы для анализа с помощью ступки и пестика до обеспечения пропускания через сито с размером отверстий 63 мкм. Измельченный материал высушивают в течение 2 ч в печи с терморегулятором, обеспечивающий температуру нагрева до (105 ± 1) °C, охлаждают в эксикаторе и взвешивают в платиновом тигле с точностью до 0,0002 г примерно 0,2 г пробы. Добавляют 4 г карбоната натрия.
Содержимое перемешивают в тигле стеклянной палочкой и нагревают газовой горелкой сначала слегка, а затем докрасна. После получения прозрачного плава выдерживают при температуре красного каления в течение 5—10 мин. Проверяют, обеспечен ли хороший контакт навески пробы с расплавленным карбонатом натрием, периодически перемешивая содержимое путем вращения тигля.
Тигель охлаждают, помещая его в холодную воду, чтобы отделить плав от его стенок. Твердый плав переводят в химический стакан вместимостью 600 см3, затем все оставшиеся частицы в тигле смывают в химический стакан. Добавляют воду в содержимое химического стакана, чтобы довести до общего объема приблизительно 200 см3. Химический стакан помещают на водяную баню с кипящей водой, оставляют там на 30 мин, дробя кусочки стеклянной палочкой с плоским концом, выравненной на одном из своих концов. Охлаждают, доводят до объема приблизительно 300 см3 водой, добавляют 20 см3 раствора борной кислоты. Постоянно перемешивая, добавляют раствор соляной кислоты до получения pH примерно 2 (по соответствующей индикаторной бумаге). Раствор, который может иметь иногда слегка опалесцирующий вид, переводят в мерную колбу вместимостью 500 см3, доводят до метки водой и перемешивают.
Если наблюдается белый осадок сульфата бария, его отстаивают перед тем, как продолжить анализ.
7.2.6.2 Контрольный опыт
Параллельно с анализом и в тех же самых условиях определения проводят контрольный опыт, используя те же количества всех реактивов, но заменяя объем анализируемого раствора равным количеством воды.
7.2.6.3 Построение градуировочного графика
а) Приготовление стандартных колориметрических растворов для спектрометрических измерений В каждый из шести химических стаканов вместимостью 600 см3 вводят объемы стандартного силикатного раствора, указанные в таблице 8.
Таблица 8
Стандартный силикатный раствор, см3 | Масса диоксида кремния, соответствующая объему, используемому для измерения, мкг |
о1) | 0 |
2 | 8 |
5 | 20 |
10 | 40 |
20 | 25 |
25 | 100 |
1) Компенсирующий раствор. | |
Добавляют 4 г карбоната натрия, доводят объем примерно до 300 см3 водой, добавляют 20 см3 раствора борной кислоты до установления значения pH примерно 2 (по соответствующей индикаторной бумаге). Раствор переводят в мерную колбу вместимостью 500 см3, доводят до метки водой и перемешивают.
б) Предварительный опыт и корректировка pH
В один из химических стаканов вместимостью 100 см3 переводят 20,0 см3 стандартного колориметрического раствора, содержащего 100,0 мкг диоксида кремния. Разбавляют примерно до 60 см3 водой и, постоянно перемешивая, устанавливают pH 1,1 добавлением раствора серной кислоты.
Записывают использованный объем раствора серной кислоты и отбрасывают раствор.
в) Цветное окрашивание
В каждую из шести мерных колб вместимостью 100 см3 отмеряют по 20 см3 растворов, полученных в соответствии с пунктом а). Разбавляют примерно до 60 см3 водой. Затем добавляют объем раствора серной кислоты, использованный в пункте б), и 10 см3 раствора молибдата. Перемешивают и оставляют в покое на 15 мин. Добавляют 5 см3 раствора винной кислоты, перемешивают и оставляют в покое на 5 мин. Затем добавляют 10 см3 раствора серной кислоты и 2 см3 аскорбиновой кислоты. Доводят до метки водой, перемешивают и оставляют в покое на 30 мин.
г) Спектрометрические измерения
Выполняют спектрометрические измерения с помощью спектрометра при длине волны, соответствующей максимальной абсорбции (примерно 795 нм), или спектрометра с соответствующим фильтром после установки прибора на нулевое значение, используя воду в качестве раствора сравнения.
д) Построение градуировочного графика
Вычитают абсорбцию компенсирующего раствора из значений абсорбции каждого стандартного колориметрического раствора. Строят график, нанося, например, на ось абсцисс массы кремния SiO2, содержащиеся в колориметрических растворах, в микрограммах, а на ось ординат — соответствующие значения абсорбции.
7.2.6.4 Определение
а) Предварительный опыт и корректировка pH
В один из химических стаканов вместимостью 100 см3 переводят 20 см3 анализируемого раствора по 7.2.6.1. Разбавляют примерно до 60 см3 водой и, постоянно перемешивая, регулируют кислотность до pH 1,1 добавлением раствора серной кислоты.
Записывают использованный объем серной кислоты и отбрасывают раствор.
б) Цветное окрашивание
В мерную колбу вместимостью 100 см3 переводят не более 20 см3 анализируемого раствора по 7.2.6.1, содержащего не более 100 мкг SiO2. Если используется количество менее 20 см3, то добавляют раствор для разбавления, чтобы получить точно общий объем в 20 см3.
Разбавляют до 60 см3 водой. Затем добавляют объем раствора серной кислоты, использованный в пункте а), и 10 см3 раствора молибдата. Перемешивают и оставляют в покое 15 мин. Добавляют 5 см3 раствора винной кислоты, перемешивают и оставляют в покое 5 мин. Затем добавляют 10 см3 раствора серной кислоты и 2 см3 аскорбиновой кислоты. Разбавляют до метки водой, перемешивают и оставляют в покое 30 мин.
в) Спектрометрические измерения
Проводят спектрометрические измерения анализируемого раствора по пункту б) и раствора контрольного опыта по 7.2.6.2 в соответствии с методом определения, указанным в 7.2.6.3, пункт г) после установки прибора на нулевое значение абсорбции, используя в качестве раствора сравнения воду.
7.2.7 Обработка результатов
По градуировочному графику по 7.2.6.3 определяют содержание диоксида кремния, соответствующее абсорбции анализируемого раствора и раствора контрольного опыта.
Массовую долю диоксида кремния WSio2> %, вычисляют по формуле
<7'3)
2 П?0
где т1 — масса диоксида кремния SiO2, определенная в аликвотной части анализируемого раствора, отобранной для цветного окрашивания, мкг;
т2 — масса диоксида кремния SiO2, определенная в соответствующей аликвотной части раствора контрольного опыта, мкг;
mQ — масса навески пробы по 7.2.6.1, г;
rD — отношение объема анализируемого раствора к объему аликвотной части, отобранной для цветного окрашивания.
7.2.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
7.2.9 Сравнение результатов, полученных с использованием различных методов
Этот метод был использован для анализа ряда проб плавиковых шпатов, содержание диоксида кремния в которых было определено гравиметрическим кремнемолибденовым методом (метод лаборатории Леверкузена) и методом Жиффорда.
Результаты приведены в таблице 9.
Таблица 9
Проба | Массовая доля диоксида кремния, % | ||
Спектрометрический метод, приведенный в настоящем стандарте | Г равиметрический кремнемолибденовый метод (среднее) | Метод Жиффорда (среднее) | |
Плавиковые шпаты по сравнительным испытаниям: 1 | 1,55; 1,56 | 1,60 | 1,5 |
2 | 1,27; 1,27 | 1,24 | 1,2 |
3 | 1,73; 1,80 | 1,70 | — |
«Ротглебероде» | 1,03; 1,03 | 0,99 | 1,0 |
Плавиковые шпаты по обычным анализам: А | 0,30; 0,31 | 0,31 | _ |
В | 0,50; 0,51 | — | 0,5 |
С | 1,09; 1,10 | — | 1,1 |
Д | 0,17; 0,17 | — | 0,2 |
Е | 1,68; 1,67 | 1,66 | — |
8 Метод определения металлических оксидов Ме2О3
8.1 Сущность метода
Настоящий раздел устанавливает гравиметрический метод определения содержания металлических оксидов Ме2О3 при массовой доле от 0,1 % до 5 %.
Метод основан на осаждении металлических оксидов Ме2О3 уротропином после отделения сульфата бария и окисления железа в растворе, полученном после обработки навески серной и фтористоводородной кислотами и сплавления остатка со смесью карбонатов калия и натрия. Осадок металлических оксидов Ме2О3 прокаливают при 900 °C — 1000 °C до постоянной массы и взвешивают.
8.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
печь электрическую муфельную, обеспечивающую температуру нагрева 1000 °C;
термопару термоэлектрическую хромель-алюмелевую ТХА по ГОСТ 6616;
чашки платиновые по ГОСТ 6563;
тигли платиновые по ГОСТ 6563;
кислоту серную по ГОСТ 4204;
кислоту соляную по ГОСТ 3118 и разбавленную 1:1 и 1:4;
кислоту фтористоводородную (плавиковую кислоту) по ГОСТ 10484;
смесь карбонатов калия и натрия, безводный по ГОСТ 4332;
водорода перекись (пергидроль) по ГОСТ 10929;
метиловый красный (4-диметиламиноазобензол-2 карбоновая кислота), 0,1 %-ный спиртовой раствор;
аммоний азотнокислый по ГОСТ 22867, раствор 20 г/дм3, нейтрализованный аммиаком до пожелтения индикатора метилового красного;
аммиак водный по ГОСТ 3760, разбавленный 1:1;
уротропин (гексаметилентетрамин) по ГОСТ 1381, раствор 250 г/дм3;
азотнокислое серебро по ГОСТ 1277, 1 %-ный раствор;
бумагу конго.
8.3 Проведение анализа
Навеску плавикового шпата массой 1 г помещают в платиновую чашку, увлажняют тремя-четырьмя каплями воды, приливают 3 см3 серной кислоты и нагревают до появления паров серной кислоты. Затем содержимое чашки охлаждают, доливают 10 см3 фтористоводородной кислоты и выпаривают до густых паров серной кислоты. Выпаривание с фтористоводородной кислотой повторяют трижды. Остаток охлаждают, обмывают стенки чашки водой, добавляют две-три капли серной кислоты и выпаривают досуха.
Далее к содержимому чашки добавляют 10 г смеси карбонатов калия и натрия и сплавляют при 850 °C — 900 °C в течение 10—15 мин. Плав выщелачивают горячей водой в стакан вместимостью 300 см3, осторожно добавляют 20 см3 соляной кислоты и 3—4 капли пергидроля. Раствор кипятят в течение 5 мин, после чего фильтруют через плотный фильтр в стакан вместимостью 600 см3. Осадок на фильтре промывают горячей водой, подкисленной несколькими каплями соляной кислоты. Общий объем фильтрата и промывных вод должен быть не менее 350—400 см3. Полученный раствор нейтрализуют раствором аммиака до начала покраснения индикаторной бумаги конго. Затем добавляют несколько капель соляной кислоты, разбавленной 1:4, до перехода цвета индикаторной бумаги конго в сиреневый цвет, приливают 15 см3 уротропина и раствор выдерживают в течение 10 мин при температуре примерно 80 °C, не доводя его до кипения. Осадок отфильтровывают на фильтр средней плотности и промывают горячим раствором азотнокислого аммония до исчезновения реакции на ион хлора (проба раствором азотнокислого серебра, подкисленного азотной кислотой).
Осадок с фильтром помещают во взвешенный платиновый тигель, сушат, озоляют и прокаливают при температуре 900 °C — 1000 °C в течение 1 ч. После охлаждения в эксикаторе тигли с осадком взвешивают. Прокаливание повторяют до постоянной массы.
8.4 Обработка результатов
8.4.1 Массовую долю металлических оксидов Ме2О3 Х^, %, вычисляют по формуле
х 100
(8.1)
1 т
где т1 — масса металлических оксидов Ме2О3, г; т — масса навески плавикового шпата, г.
8.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 10.
Таблица 10
Массовая доля металлических оксидов Ме2О3, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,10 до 0,30 включ. | 0,07 | 0,10 |
Св. 0,30 » 1,00 » | 0,12 | 0,15 |
» 1,00 » 3,00 » | 0,15 | 0,20 |
» 3,00 » 5,00 » | 0,20 | 0,25 |
9 Методы определения железа
9.1 Спектрофотометрический метод
9.1.1 Сущность методаНастоящий подраздел устанавливает спектрофотометрический метод определения массовой доли железа при его массовой доле 0,05 % до 5 %.
Метод основан на образовании в аммиачной среде окрашенного в желтый цвет комплексного соединения ионов железа с сульфосалициловой кислотой.
9.1.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
спектрофотометр или фотоэлектроколориметр любого типа для измерения в видимой области спектра;
кислоту соляную по ГОСТ 3118;
кислоту азотную по ГОСТ 4461;
аммиак водный по ГОСТ 3760;
кислоту сульфосалициловую по ГОСТ 4478, раствор 200 г/дм3;
кислоту серную по ГОСТ 4204, растворы 160 г/дм3 и 0,0005 моль/дм3;
соль Мора по ГОСТ 4208;
стандартные растворы железа:
раствор А, приготовленный следующим образом: 0,702 г соли Мора растворяют в воде, содержащей 0,4 см3 16 %-ного раствора серной кислоты, доводят объем раствора водой до 100 см3 и перемешивают. Раствор применяют свежеприготовленным. 1 см3 раствора А содержит 1 мг железа;
раствор Б, приготовленный следующим образом: 10 см3 раствора А помещают в мерную колбу вместимостью 1000 см3, доводят объем раствора до метки 0,0005 моль/дм3 раствором серной кислоты и перемешивают. 1 см3 раствора Б содержит 0,01 мг железа.
9.1.3 Проведение анализа
9.1.3.1 Навеску плавикового шпата массой 0,5 г помещают в коническую колбу вместимостью 250 см3, увлажняют несколькими каплями воды, приливают 20 см3 соляной кислоты, нагревают 10 мин (не доводя до кипения). Затем добавляют 10 см3 азотной кислоты и выпаривают досуха, избегая перегревания. Приливают 10 см3 соляной кислоты и вновь выпаривают досуха. Выпаривание с соляной кислотой повторяют дважды.
Сухой остаток смачивают 5 см3 соляной кислоты, приливают 20 см3 горячей воды и кипятят 5 мин, накрыв часовым стеклом, до растворения растворимых солей.
Нерастворимый остаток отфильтровывают на плотный фильтр, промывают три-четыре раза горячей водой, подкисленной соляной кислотой, и пять-шесть раз горячей водой, собирая фильтрат и промывные воды в мерную колбу вместимостью 200 см3. Раствор охлаждают, доводят до метки водой и перемешивают.
Аликвотную часть раствора 20 см3 (при содержании железа 0,1 %) или 5 см3 (при содержании железа от 0,1 % до 0,5 %) помещают в мерную колбу вместимостью 100 см3, приливают 10 см3 сульфосалициловой кислоты, добавляют аммиак до окрашивания раствора в желтый цвет и в избыток 2,0 см3, доливают водой до метки и перемешивают. Измеряют оптическую плотность раствора, применяя светофильтр с максимумом светопропускания 424 нм в кювете толщиной колориметрируемого слоя 50 мм.
Раствором сравнения служит раствор контрольного опыта, проведенный через все стадии анализа. По величине оптической плотности испытуемого раствора находят содержание железа по градуировочному графику.
9.1.3.2 Для построения градуировочного графика в 10 мерных колб вместимостью по 100 см3 отмеривают бюреткой 1; 2; 3; 4; 5; 6; 7; 8; 9 и 10 см3 стандартного раствора Б. В одиннадцатую колбу стандартный раствор не отмеривают. В каждую колбу добавляют по 10 см3 сульфосалициловой кислоты и далее продолжают анализ, как указано в 9.1.3.1.
Раствором сравнения служит раствор одиннадцатой колбы, который не содержит стандартный раствор железа.
По полученным значениям оптической плотности растворов и известным содержаниям железа строят градуировочный график.
9.1.4 Обработка результатов
9.1.4.1 Массовую долю железа WFe, %, вычисляют по формуле
... mj-VMOO
Fe Ц.т-1000’ (9'1)
где П71 — масса железа, найденная по градуировочнаму графику, мг;
V — объем испытуемого раствора в мерной колбе, см3;
V1 — объем аликвотной части испытуемого раствора, см3;
т — масса навески плавикового шпата, г.
9.1.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 11.
Таблица 11
Массовая доля железа, % | Допускаемые расхождения, % | |||||
предел повторяемости г | предел воспроизводимости R | |||||
От | 0,050 | ДО | 0,100 | включ. | 0,007 | 0,010 |
Св. | 0,100 | » | 0,300 | » | 0,012 | 0,015 |
» | 0,300 | » | 0,500 | » | 0,015 | 0,020 |
9.2 Атомно-абсорбционный метод
9.2.1 Сущность метода
Настоящий подраздел устанавливает атомно-абсорбционный метод определения массовой доли железа при его массовой доле 0,05 % до 5 %.
Метод основан на измерении поглощения линии железа 248,3 нм при введении растворов проб и эталонных растворов в воздушно-пропан-бутановое пламя. Пробы плавикового шпата предварительно переводят в раствор кислотным разложением.
9.2.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
спектрофотометр атомно-абсорбционный любой марки (пламенный вариант);
лампу с полым катодом (ЛПК), излучающую спектр атомов железа;
кислоту азотную по ГОСТ 4461;
кислоту соляную по ГОСТ 3118;
хлорид кальция, раствор готовят следующим образом: 2,96 г хлорида кальция помещают в мерную колбу вместимостью 1000 см3, доводят до метки водой и перемешивают;
газ пропан-бутан по ГОСТ 20448;
серную кислоту по ГОСТ 4204, растворы 160 г/дм3 и 0,0005 моль/дм3;
соль Мора по ГОСТ 4208;
стандартные растворы железа по 9.2.3.
9.2.3 Подготовка к проведению испытания
Стандартные растворы железа готовят следующим образом:
раствор А: 0,702 г соли Мора растворяют в воде, содержащей 0,4 см3 10 %-ного раствора серной кислоты, приливают воду до 100 см3 и перемешивают. Раствор применяют свежеприготовленный, 1 см3 раствора А содержит 1 мг железа;
раствор Б: 10 см3 раствора А помещают в мерную колбу вместимостью 100 см3, доливают до метки 0,0005 моль/дм3 раствором серной кислоты и перемешивают. 1 см3 раствора Б содержит 0,1 мг железа.
Серию стандартных растворов готовят следующим образом: в шесть мерных колб вместимостью по 100 см3 отмеривают 0,5; 1,0; 2,0; 4,0; 5,0 и 10,0 см3 раствора Б, до метки доливают раствором хлорида кальция, что соответствует 0,5; 1,0; 2,0; 4,0; 5,0; 10,0 мкг/см3 железа в конечном объеме. Растворы тщательно перемешивают. Фотометрируют в режиме работы прибора Перкин-Элмер 303 или 403 согласно таблице 12.
Таблица 12
Параметры | Оптимальные условия определения железа |
Длина волны, нм Спектральная ширина щели, А Величина тока ЛПК со спектром железа, мА Давление воздуха, Па Расход воздуха, дм3/мин Расход пропан-бутан, дм3/мин Длина пламени, см Высота прохождения света над горелкой, см | 249,3 2 30 1,8 • 105 (1,8 кгс/см3) 16 1 9—12 1 |
Оптимальные условия уточняются в зависимости от конкретного типа атомно-абсорбционного спектрофотометра.
9.2.4 Проведение анализа
Навеску плавикового шпата массой 1 г при содержании железа до 0,1 % и 0,5 г при содержании железа от 0,1 % до 0,5 % помещают в коническую колбу вместимостью 250 см3, смачивают несколькими каплями воды, приливают 20 см3 соляной кислоты, нагревают 10 мин (не доводя до кипения). Затем добавляют 10 см3 азотной кислоты и выпаривают досуха, избегая перегревания. Далее приливают 10 см3 соляной кислоты и вновь выпаривают досуха. Выпаривание с соляной кислотой повторяют дважды, приливая каждый раз по 10 см3 кислоты. Сухой остаток смачивают 5 см3 соляной кислоты, приливают 20 см3 горячей воды, кипятят 5 мин, накрыв колбу часовым стеклом, до растворения растворимых солей. Содержимое охлаждают и переводят в мерную колбу вместимостью 200 см3, доводят объем раствора до метки водой и перемешивают.
Растворы проб и эталонов распыляют в пламени и измеряют поглощение линии железа. Измерения проводят по методу ограничивающих растворов, при котором стандартные растворы подбирают так, чтобы поглощение для одного было меньше, а для другого больше по сравнению с поглощением для раствора пробы.
На спектрофотометрах, имеющих режим измерения «Концентрация» (например, фирмы Перкин-Элмер и др.), используют метод трех эталонов. Измерения для каждой пробы проводят три раза. По среднему результату измерений для метода ограничивающих растворов определяют концентрацию железа в растворе в мкг/см3.
9.2.5 Обработка результатов
9.2.5.1 Массовую долю железа l/l/Fe, %, вычисляют по формуле
/п-106 (9.2)
где т1 — масса железа, найденная по градуировочному графику, мкг/см3;
V — объем испытуемого раствора в мерной колбе, см3;
т — масса навески пробы, г.
9.2.5.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 13.
Таблица 13
Массовая доля железа, % | Допускаемые расхождения, % | |||||
предел повторяемости г | предел воспроизводимости R | |||||
От | 0,050 | ДО | 0,100 | включ. | 0,007 | 0,010 |
Св. | 0,100 | » | 0,300 | » | 0,012 | 0,015 |
» | 0,300 | » | 0,500 | » | 0,015 | 0,020 |
9.3 Спектрометрический метод определения содержания железа
9.3.1 Назначение методаНастоящий подраздел устанавливает спектрометрический метод определения содержания железа с 1,10-фенантролином в плавиковом шпате для производства керамики.
Метод распространяется на материалы с содержанием железа, выраженного как Fe2O3, от 0,1 % до 2,0 % (по массе).
9.3.2 Сущность метода
Щелочное сплавление навески со смесью карбоната натрия и борной кислоты.
Растворение плава в избытке соляной кислоты.
Восстановление железа (III) соляно-кислым гидроксиламином.
Образование железа (III) — 1,10-фенантролинового комплекса в буферной среде (pH между 3 и 5). Спектрометрическое измерение окрашенного комплекса при длине волны 510 нм.
9.3.3 Реактивы
В процессе проведения анализа используют реактивы аналитической степени чистоты и дистиллированную воду или воду эквивалентной кислоты.
Натрия карбонат безводный.
Кислота борная.
Кислота соляная, разбавленная 1:1.
Разбавляют 1 объем соляной кислоты р = 1,18 г/см3 равным объемом воды.
Гидроксиламин соляно-кислый HONH3CI, раствор 10 г/дм3.
1,1- фенантролин моногидрат C12H8N2 • ЗН2О, раствор 2, г/дм3.
Натрия ацетат тригидрат CH3COONa ■ ЗН2О, раствор 500 г/дм3.
Железо, стандартный раствор, соответствующий 0,100 г Fe2O3 на 1 дм3. Взвешивают с точностью до 1 мг 0,605 г железоаммонийных квасцов 24-водных [Fe2(SO4)3(NH4)2SO4 • 24Н2О] помещают в химический стакан и растворяют в воде. Добавляют 10 см3 серной кислоты р ~ 1,84 г/дм3, охлаждают, количественно переносят раствор в мерную колбу вместимостью 1000 см3, разбавляют водой до метки и перемешивают.
1 см3 такого стандартного раствора содержит 0,100 мг.
9.3.4 Оборудование
Для проведения анализа применяют:
электропечь с устройством для регулирования температуры (105 ± 2) °C;
платиновую чашку с плоским дном, диаметром 80 мм и глубиной 35 мм, с платиновой крышкой;
муфельную печь с режимом работы до 1000 °C;
молекулярно-абсорбционный спектрометр, снабженный оптическими кюветами с длиной оптического пути 20 мм.
9.3.5 Проба для анализа
В качестве пробы для анализа используют остаток от определения потери массы при 105 °C.
Примечание — Стандарт для плавикового шпата, предназначенного для производства плавиковой кислоты, равно применим к плавиковому шпату для производства керамики.
9.3.6 Проведение анализа
9.3.6.1 Навеска и приготовление раствора для анализа
Истирают несколько граммов для анализа пробы по 9.3.5 в агатовой ступке до прохождения частиц через сито с размером отверстий 63 мкм. Высушивают истертый материал в течение 2 ч в печи при температуре (105 ± 2) °C, охлаждают в эксикаторе и взвешивают с точностью до 1 мг примерно 0,5 г этой пробы в платиновую чашку.
Добавляют 6 г борной кислоты, 4 г безводного карбоната натрия и тщательно перемешивают предпочтительно платиновой палочкой. Накрывают чашку крышкой, помещают на жаропрочную и термоизоляционную плиту, сначала слабо нагревают и затем увеличивают температуру постепенно до ослабления реакции.
Переносят в муфельную печь, нагретую до температуры примерно 1000 °C и продолжают нагрев до полного сплавления.
Удаляют чашку из муфельной печи и дают остыть на воздухе. Добавляют горячую воду в чашку, нагревают на водяной бане до растворения плава, подкисляют, добавляя осторожно 20 см3 соляной кислоты, и количественно переносят в мерную колбу вместимостью 250 см3. Охлаждают, разбавляют водой до метки и перемешивают.
9.3.6.2 Контрольный опыт
Одновременно с определением выполняют контрольный опыт, следуя той же методике и используя такие же количества реактивов, что и при определении, исключив навеску.
9.3.6.3 Приготовление градуировочного графика
а) Приготовление градировочных растворов.
В каждую из семи мерных колб вместимостью по 250 см3 отмеривают объемы стандартного раствора железа в соответствии с таблицей 14.
б) Образование абсорбционного комплекса.
Добавляют к содержимому каждой колбы воду до 200 см3, затем добавляют 5 см3 раствора соляно-кислого гидроксиламина, перемешивают одну минуту. Добавляют 10 см3 раствора ацетата натрия, перемешивают 1 мин. Добавляют 10 см3 раствора ацетата натрия и 5 см3 раствора 1,10-фенантролина, разбавляют водой до метки и перемешивают.
Таблица 14
Объем стандартного раствора железа (9.3.3), см3 | Соответствующая масса, мг |
о1) | 0 |
1,0 | 0,1 |
2,0 | 0,2 |
3,0 | 0,3 |
4,0 | 0,4 |
5,0 | 0,5 |
6,0 | 0,6 |
1> Контрольный опыт на реактивы для градуировки. |
в) Спектрометрические измерения
Через 15 мин выполняют спектрометрические измерения градуировочных растворов, используя спектрометр, настроенный на волну примерно 510 нм, после регулирования прибора на ноль абсорбции, используя в качестве раствора сравнения воду.
г) Построение градуировочного графика
Вычитают значение абсорбции градуировочного компенсирующего раствора (см. таблицу 14) из значения абсорбции каждого градуировочного раствора для получения чистой абсорбции.
Строят градуировочный график, откладывая, например, значения массы (в миллиграммах) оксида железа (III), содержащегося в 250 см3 градуировочных растворов, на оси абсцисс, а соответствующие значения чистой абсорбции — на оси ординат.
9.3.6.4 Определение
а) Аликвотная порция раствора для испытаний
В соответствии с установленным содержанием железа помещают аликвотную порцию раствора для испытаний (см. 9.3.6.1), как указано в таблице 15, в мерную колбу вместимостью 250 см3.
Таблица 15
Массовая доля Fe2O3, % | Аликвотная порция, см3 | |||
От | 0,1 | ДО | 0,5 включ. | 50 |
Св. | 0,5 | » | 1,0 » | 20 |
» | 1,0 | » | 2,0 » | 10 |
б) Образование абсорбционного комплекса
К аликвотной порции раствора для анализа в мерной колбе вместимостью 250 см3 добавляют такие же количества всех реактивов, какие добавлялись к стандартному раствору железа, разбавляют водой до метки и перемешивают.
в) Спектрометрические измерения
Через 15 мин выполняют спектрометрические измерения раствора для испытаний и контрольного раствора, следуя методике, установленной в п.9.3.6.3, после регулирования прибора на нуль абсорбции, используя в качестве раствора сравнения воду.
9.3.7 Обработка результатов
Используя градуировочный график (см 9.3.6.3), определяют массу железа, соответствующие значениям абсорбции раствора для анализа и контрольного раствора.
Массовую долю железа WFe, %, вычисляют по формуле
ЛТо-10
(9.3)
где т0 — масса навески (9.3.6.1), г;
т1 — масса оксида железа (III), определенная в аликвотной порции раствора для анализа, мг;
т2 — масса оксида железа (III), определенная в аликвотной порции раствора контрольного опыта, мг;
rD — отношение объема раствора для анализа к объему аликвотной порции, отобранной для определения.
9.3.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
10 Метод определения серы (общей)
10.1 Сущность метода
Настоящий раздел устанавливает титриметрический метод определения серы (общей) при массовой доле от 0,05 % до 5 %.
Метод основан на сжигании навески в токе углекислоты при температуре 1200 °C — 1250 °C. Образовавшийся диоксид серы выносится током углекислого газа в адсорбционный сосуд и поглощается в нем водой с образованием сернистой кислоты, которую оттитровывают раствором йода в присутствии индикатора-крахмала. Сжигание серы в атмосфере углекислоты происходит стехиометрически, что позволяет пользоваться теоретическим титром раствора йода.
10.2 Общие требования
Достоверность получаемых результатов анализа контролируется путем одновременного проведения анализа на содержание серы в стандартном образце флюоритового концентрата № 1822-80 или ему подобного.
10.3 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
Установку для определения содержания серы (см. рисунок 4).
1 — баллон с углекислотой, снабженный редукционным вентилем для пуска и регулирования тока углекислого газа; 2, 3 — склянки Тищенко; 4 — сушильная колонка; 5 — горизонтальная трубчатая печь с силитовыми нагревателями; б — огнеупорная муллиткремнеземистая трубка; 7 — грушеобразная трубка; 8 — поглотительный сосуд; 9 — барбатер;
10 — бюретка
Рисунок 4 — Установка для определения содержания серы
Установка состоит из следующих элементов: баллона с углекислотой 1, снабженного редукционным вентилем для пуска и регулирования тока углекислого газа; склянки Тищенко 2, содержащей 5 %-ный раствор сульфата меди; склянки Тищенко 3, содержащей 4 %-ный раствор марганцовокислого калия в 30 %-ном растворе гидроксида калия; сушильной колонки 4, заполненной прокаленным хлоридом кальция; горизонтальной трубчатой печи 5 с силитовыми нагревателями, обеспечивающими нагревание до температуры (1300 ± 20) °C; термопары платинородиевой с терморегулятором; трансформатора и амперметра для регулировки питания печи; огнеупорных муллиткремнеземистых трубок 6 длиной 650—750 мм с внутренним диаметром 20—22 мм. Перед применением трубки (новые) должны быть прокалены при рабочей температуре по всей длине. Трубки с обеих сторон закрывают хорошо подогнанными резиновыми пробками. В отверстия пробок вставляются стеклянные или латунные трубки. Для предотвращения обгорания резиновых пробок внутреннюю торцовую поверхность закрывают асбестовыми прокладками; грушеобразной трубки 7, заполненной стекловатой, служащей для улавливания твердых частиц. Газообразные продукты сжигания через барбатер 9 поступают в поглотительный сосуд 8. Барбатер должен быть изготовлен из оргстекла или фторопласта, быть разборным и состоять из двух частей. Для титрования используется бюретка 10 на 25 см3;
лодочки муллиткремнеземистые, прокаленные при температуре 1000 °C;
крючок из жаропрочной проволоки;
гидроксид калия, раствор 300 г/дм3;
оксид алюминия безводный, хорошо промытый водой и прокаленный при температуре 1000 °C в течение 5 ч;
перманганат калия по ГОСТ 20490, 4 %-ный раствор в 30 %-ном растворе гидроксида калия; сульфат меди 5-водный по ГОСТ 4165, раствор 50 г/дм3;
хлорид кальция плавленый;
стекловату, хорошо промытую водой и высушенную;
крахмал растворимый по ГОСТ 10163, 0,5 %-ный раствор; готовят следующим образом: 5 г растворимого крахмала растирают в ступке с 50 см3 холодной воды, вливают тонкой струйкой в колбу, где находится 700 см3 горячей воды. Раствор кипятят 2—3 мин, охлаждают, разбавляют холодной водой до 1000 см3;
титрованные растворы йода с концентрациями 0,025 и 0,005 моль/дм3 готовят из фиксанала 0,05 моль/дм3 соответствующим разбавлением. Растворы хранят в склянке из темного стекла. Раствор 0,005 моль/дм3 йода готовят в день употребления. В случае длительного хранения титрованного раствора титр Т проверяют по стандартному образцу СО К-3 № 1822-80 состава флюоритового концентрата и рассчитывают по формуле
_ Ст
“у.100’ (10.1) где С — массовая доля серы (общей) в стандартном образце, %;
т — масса навески стандартного образца, г;
V — объем раствора йода, израсходованный на титрование, см3.
10.4 Подготовка к проведению испытания
Перед началом работы нагревают печь до температуры 1250 °C. Для проверки правильности работы установки сжигают две-три навески стандартного образца флюорита по методике, приведенной в 10.5. Скорость пропускания углекислого газа 90—100 пузырьков в минуту.
Если сера представлена в виде барита, то навеску анализируемого материала перед сжиганием смешивают в лодочке с двукратным количеством оксида алюминия.
10.5 Проведение анализа
Навеску плавикового шпата массой 0,5 г при массовой доле 0,5 % серы и массой 0,1 г при массовой доле свыше 0,5 % помещают в лодочку. В поглотительный сосуд заранее наливают 150—200 см3 воды, приливают 5 см3 крахмала и перемешивают током углекислоты. Лодочку с навеской вводят в фарфоровую трубку (со стороны подачи углекислого газа) при помощи крючка в наиболее разогретый участок трубки, тотчас закрывают трубку и пускают ток углекислого газа. Прокаливание продолжают 5 мин. Титруют раствором йода 0,005 или 0,025 моль/дм3 соответственно навеске до устойчивой бледно-голубой окраски раствора.
Окончив титрование, крючком извлекают из печи лодочку, стараясь при этом не загрязнить фарфоровую трубку остатком навески. Затем в поглотительный сосуд, промытый водой, наливают новую порцию воды и крахмала.
10.6 Обработка результатов
10.6.1 Массовую долю серы (общей) l/l/s , %, вычисляют по формуле где V — объем раствора йода, израсходованный на титрование, см3;
Т — титр раствора йода по сере;
т — масса навески плавикового шпата, г.
10.6.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р = 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 16.
Таблица 16
Массовая доля серы, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,500 до 0,100 включ. | 0,007 | 0,010 |
Св. 0,100 » 0,300 » | 0,015 | 0,020 |
» 0,300 » 0,500 » | 0,03 | 0,04 |
» 0,500 » 1,500 » | 0,07 | 0,10 |
» 1,500 » 5,000 » | 0,12 | 0,15 |
11 Методы определения серы (сульфидной)
11.1 Объемный метод определения серы (сульфидной)
11.1.1 Сущность методаНастоящий подраздел устанавливает объемный метод определения серы (сульфидной) при массовой доле от 0,05 % до 0,5 %.
Метод основан на разложении сульфидов и дисульфидов соляной кислотой. Выделившийся сернистый водород поглощают раствором ацетата кадмия с образованием сульфида кадмия. Последний титруют раствором йода, избыток которого затем оттитровывают раствором тиосульфата натрия.
11.1.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
установку для определения содержания серы (сульфидной) (см. рисунок 5);
1 — аппарат Киппа для получения углекислого газа; 2 — склянка Тищенко с раствором сульфата меди; 3 — склянка Тищенко с водой; 4 — капельная воронка; 5 — колба вместимостью 500 см3 для разложения навески; б — колба для улавливания паров соляной кислоты; 7 — первый приемник; 8— второй приемник
Рисунок 5 — Установка для определения содержания серы (сульфидной)
кислоту серную по ГОСТ 4204, разбавленную 1:9;
кислоту соляную по ГОСТ 3118, разбавленную 1:1 и 1:5;
кислоту уксусную по ГОСТ 61;
титрованный раствор йода, 0,025 моль/дм3; готовят из фиксанала 0,05 моль/дм3 соответствующим разбавлением. Раствор хранят в склянке из темного стекла;
раствор тиосульфата натрия 0,05 моль/дм3 готовят из фиксанала 0,1 моль/дм3 соответствующим разбавлением. Раствор хранят в склянке из темного стекла;
титрованный раствор дихромата калия 0,01 моль/дм3; готовят следующим образом: 2,9422 г дихромата калия растворяют в воде и разбавляют до 1000 см3 водой;
калий йодистый по ГОСТ 4232, свежеприготовленный раствор;
крахмал растворимый по ГОСТ 10163, 1 %-ный свежеприготовленный раствор;
кадмий уксуснокислый, раствор; готовят следующим образом: 50 г соли и 10 см3 уксусной кислоты растворяют в 1000 см3 воды;
оксид хрома по ГОСТ 3776;
цинк гранулированный;
спирт этиловый гидролизный ректификованный;
сульфат меди 5-водный по ГОСТ 4165, раствор 40 г/дм3.
11.1.3 Проведение анализа
11.1.3.1 Навеску плавикового шпата массой 3—5 г (в зависимости от содержания сульфидной серы) помещают в колбу для разложения навески, смачивают несколькими каплями этилового спирта, добавляют 0,1 г оксида хрома и 5 г цинка. В колбу для улавливания паров соляной кислоты наливают 100 см3 воды, затем в первый приемник наливают 50 см3 раствора уксуснокислого кадмия и 50 см3 воды, а во второй приемник — 25 см3 раствора ацетата кадмия и 25 см3 воды. Установку собирают, как указано на рисунке 5.
В капельную воронку наливают 30 см3 разбавленной 1:5 соляной кислоты и, приливая по каплям кислоту из капельной воронки в колбу, разлагают навеску пробы. После того как вся кислота будет израсходована, через всю установку пропускают ток углекислого газа со скоростью два-три пузырька в секунду и нагревают содержимое колбы в течение 10 мин. Далее отключают приемники. Во втором приемнике должно быть лишь незначительное помутнение раствора ацетата кадмия, в противном случае анализ повторяют с меньшей навеской.
Содержимое первого и второго приемников объединяют, приливают 25 см3 0,025 моль/дм3 раствора йода (при этом должен остаться избыток йода, в противном случае количество добавляемого раствора йода увеличивают), затем 20 см3 разбавленной 1:1 соляной кислоты, 2—3 см3 раствора крахмала и титруют раствором тиосульфата натрия до исчезновения синей окраски.
11.1.3.2 Для определения количества 0,05 н. раствора тиосульфата натрия, израсходованного на титрование 25 см3 (или более) 0,025 моль/дм3 раствора йода, отбирают 25 см3 (или более) 0,025 моль/дм3 раствора йода в колбу вместимостью 300 см3, добавляют 20—30 см3 воды и титруют 0,05 н. раствором тиосульфата натрия до окрашивания раствора в желтый цвет. Затем прибавляют 2—3 см3 раствора крахмала и продолжают титрование до полного обесцвечивания раствора.
11.1.3.3 Для установки титра 0,05 н. раствора тиосульфата натрия пипеткой отбирают 25 см3 0,05 н. раствора дихромата калия в колбу для титрования, в которую налито 10—12 см3 разбавленной 1:9 серной кислоты, 5—7 см3 раствора йодида калия, и выдерживают 5—7 мин в темном месте. Затем раствор доливают водой до 200 см3 и титруют раствором тиосульфата натрия до окрашивания раствора в светло-желтый цвет. Далее приливают 2—3 см3 раствора крахмала и продолжают титрование тиосульфата до перехода синей окраски раствора в светло-зеленую.
Титр 0,05 н. раствора тиосульфата натрия Т по сере вычисляют по формуле
_ 0,000962 V
Т =----ц----• (11.1)
где 0,000962 — титр 0,05 н. раствора дихромата калия по сере;
V — объем 0,05 н. раствора дихромата калия, см3;
V1 — объем 0,05 н. раствора тиосульфата натрия, израсходованный на титрование, см3.
11.1.4 Обработка результатов
11.1.4.1 Массовую долю серы (сульфидной) W/s2-, %, вычисляют по формуле где Т — титр 0,05 н. раствора тиосульфата натрия по сере;
V — объем 0,05 н. раствора тиосульфата натрия, израсходованный на титрование 25 см3 (или более) 0,025 моль/дм3 раствора йода, см3;
— объем 0,05 н. раствора тиосульфата натрия, израсходованный на титрование избытка раствора йода, см3;
m — масса навески плавикового шпата, г.
11.1.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р= 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 17.
Таблица 17
Массовая доля серы, % | Допускаемые расхождения, % | |
предел повторяемости г | предел воспроизводимости R | |
От 0,050 до 0,100 включ. | 0,007 | 0,010 |
Св. 0,100 » 0,300 » | 0,015 | 0,020 |
» 0,30 » 0,50 » | 0,03 | 0,04 |
11.2 Йодометрический метод определения содержания сульфида
11.2.1 Назначение методаНастоящий подраздел устанавливает йодометрический метод определения сульфида в плавиковом шпате для производства плавиковой кислоты и керамики.
Метод применим к материалам с содержанием сульфида, выраженного в виде серы, более или равным 0,001 % (по массе).
Примечание — Плавиковый шпат для производства плавиковой кислоты и керамики обычно не содержит полисульфидов.
Метод не применим, если предполагается их присутствие.
11.2.2 Сущность метода
Разложение навески в закрытом аппарате в смеси растворов соляной кислоты, хлорида олова (II) и борной кислоты. Поглощение высвободившегося сероводорода, переносимого потоком аргона или азота, не содержащих кислорода в растворе ацетата цинка, и йодометрическое определение образовавшегося сульфида цинка.
11.2.3 Реактивы
При анализе используют реактивы только аналитической чистоты и только дистиллированную воду либо воду эквивалентной чистоты.
Кислота борная.
Азот или аргон, не содержащие кислород.
Примечание — При подозрении на присутствие кислорода газ сначала пропускают через промывную склянку, содержащую щелочной раствор пирогаллола.
Кислота соляная, раствор. Разбавляют один объем соляной кислоты -1,18 г/см двумя объемами воды.
Олова хлорид (II), раствор 200 г/дм3. Растворяют 200 г дигидрата хлорида олова (II) SnCI2 • 2Н2О в 300 см3 соляной кислоты, с плотностью 1,18 г/см3, и разбавляют водой до 1000 см3.
Цинка ацетат, раствор 30 г/дм3. Растворяют 30 г дигидрата ацетата цинка и 6 см3 ледяной уксусной кислоты в 1000 см3 воды.
= 0,005 моль/дм3. Важно, чтобы этот раствор
Йод, стандартный раствор с концентрацией с
был свежеприготовленным, полученным разбавлением стандартного раствора йода с концентрацией
с
= 0,05 моль/дм3.
Натрия тиосульфат, стандартный раствор Na2S2O3 с концентрацией 0,01 моль/дм3. Важно, чтобы этот раствор был свежеприготовленным, полученным разбавлением стандартного раствора тиосульфата натрия Na2S2O3 с концентрацией 0,10 моль/дм3.
Раствор крахмала. 1 г крахмала растворяют в 10 см3 воды и медленно приливают суспензию к 200 см3 кипящей воды. Кипятят в течение 1 мин. Охлаждают и фильтруют в колбу со стеклянной крышкой.
11.2.4 Оборудование
Электрическая печь с терморегулятором, позволяющим контролировать температуру в призмах (105 ± 2) °C.
Аппарат преобразования и поглощения газа, содержащий промывную склянку; колба с плоским дном, снабженная капельной воронкой и обратным холодильником.
Примечание — Пример типичного аппарата показан на рисунке 6.
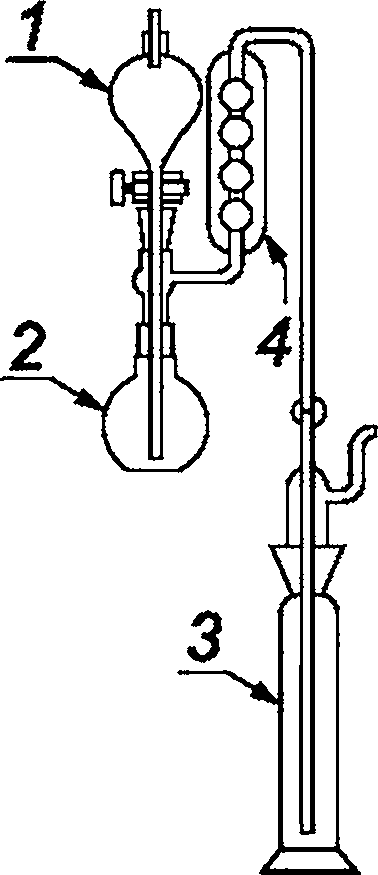
1 — капельная воронка; 2 — колба с плоским дном; 3 — промывная склянка; 4 — обратный холодильник
Рисунок 6 — Пример аппарата преобразования и поглощения газа
11.2.5 Проба для анализа
Для приготовления пробы для анализа используют остаток от определения потери массы при прокаливании при 105 °C.
Примечание — ГОСТ 7618, применяемый для плавикового шпата для производства плавиковой кислоты, в равной степени применим к плавиковому шпату для производства керамики.
11.2.6 Проведение анализа
11.2.6.1 Навеска
Истирают несколько граммов пробы для испытания по 11.2.5 в агатовой ступке до размера частиц, проходящих через сито с отверстиями 63 мкм. Высушивают просеянный материал в течение 2 ч в печи при температуре (105 ± 2) °C.
Охлаждают в эксикаторе. Взвешивают с погрешностью до 1 мг примерно 3 г этой пробы.
Примечание — Общее содержание сульфида, выраженного в виде серы, в навеске не должно превышать 0,8 мг. Для проб, содержащих более 0,03 % (по массе) серы, масса навески должна быть пропорционально сокращена.
11.2.6.2 Контрольный опыт
Одновременно с определением проводят контрольный опыт, следуя той же процедуре и используя те же реактивы, что и при определении, но без навески.
11.2.6.3 Определение
Помещают в промывную склянку 50 см3 раствора ацетата цинка. Помещают навеску по 11.2.6.1 в колбу с плоским дном, добавляют 3 см3 борной кислоты и собирают аппарат.
Приливают смесь из 50 см3 раствора соляной кислоты и 10 см3 раствора хлорида олова (II) через капельную воронку. В горлышко воронки вставляют коробку с одним отверстием, снабженную стеклянной трубкой, и пропускают поток азота или аргона через аппарат со скоростью 50 см3/мин в течение 15 мин. Слабо кипятят содержимое колбы в течение 1 ч, не прерывая подачу газа, затем отсоединяют промывную склянку от аппарата. Убирают трубку, через которую подается газ из промывной склянки, и быстро добавляют 10 см3 йода и 8—10 см3 раствора соляной кислоты. Сразу же погружают трубку, через которую подается газ в промывную склянку, закрывают трубки, через которые подается и выходит газ, и оставляют на 10 мин. Затем удаляют пробки и тщательно промывают трубку, через которую подавался газ, собирая промывные воды в склянку. Необходимо следить за тем, чтобы все количество сульфида цинка, прилипшего к стенкам входной трубки, было растворено полностью. Проводят обратное титрование не вступившего в реакцию йода раствором тиосульфата натрия, приливая 1 см3 раствора крахмала непосредственно перед достижением конечной точки титрования.
11.2.7 Обработка результатов
Массовую долю сульфида l/l/s2-, %, вычисляют по формуле
(1о.о-ц)-(ю.о-уо) 1Q0 ft00Q16=^-у0016 т т
(11-3)
где Уо — объем раствора тиосульфата натрия, использованный в контрольном опыте, см3;
— объем раствора тиосульфата натрия, использованный при определении, см3;
т — масса навески по 11.2.6.1, г;
10,0 — объем раствора йода, добавляемый в промывную склянку, см3;
0,00016 — масса серы, соответствующая 1,00 см3 раствора тиосульфата натрия Na2S2O3 с концентрацией 0,010 моль/дм3, г.
Примечание — Если концентрации используемых стандартных растворов не соответствуют концентрациям растворов, указанных в списке реактивов, следует ввести необходимые поправки.
11.2.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
12 Методы определения фосфора
12.1 Спектрофотометрический метод определения фосфора
12.1.1 Сущность методаНастоящий подраздел устанавливает спектрофотометрический метод определения фосфора при массовой доле от 0,005 % до 0,5 %.
Метод основан на образовании в кислой среде комплекса фосфорно-молибденовой кислоты и последующем восстановлении тиомочевиной до комплексного соединения, окрашенного в синий цвет (окраска устойчивая 1—2 ч).
12.1.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
спектрофотометр или фотоэлектроколориметр любого типа для измерения в видимой области спектра;
кислоту соляную по ГОСТ 3118, разбавленную 1:3;
кислоту серную по ГОСТ 4204, разбавленную 1:5;
кислоту борную по ГОСТ 9656;
сульфат меди по ГОСТ 4165, 10 г/дм3;
тиомочевину по ГОСТ 6344, свежеприготовленный раствор 100 г/см3;
молибдат аммония по ГОСТ 3765, свежеприготовленный раствор 50 г/дм3;
ортофосфат калия однозамещенный по ГОСТ 4198; готовят следующим образом: 2 г однозамещенного фосфорнокислого калия, высушенного в эксикаторе над серной кислотой в течение 2 суток, прокаливают во взвешенном платиновом тигле, постепенно повышая температуру до 700 °C. Прокаливание повторяют до постоянной массы. Если при проверке потеря массы составила от 13,15 % до 13,40 %, то взятый реактив соответствует ГОСТ 4198, квалификации химически чистый (х. ч.);
смешанный реактив из расчета на одну пробу; готовят следующим образом: смешивают последовательно 4 см3 серной кислоты, 1 см3 раствора сульфата меди, 5 см3 раствора тиомочевины и 4 см3 раствора молибдата аммония;
стандартные растворы фосфора:
раствор А, приготовленный следующим образом: 4,393 г ортофосфата калия однозамещенного, высушенного до постоянной массы при 100 °C—105 °C, растворяют в воде и доливают водой до 1 дм3. 1 см3 раствора А содержит 1 мг фосфора. Раствор хранят в полиэтиленовой посуде;
раствор Б, приготовленный следующим образом: отбирают пипеткой 10 см3 раствора А в мерную колбу вместимостью 1000 см3, доливают до метки водой и перемешивают. 1 см3 раствора Б содержит 0,01 мг фосфора.
12.1.3 Проведение анализа
12.1.3.1 Навеску плавикового шпата массой 1 г помещают в коническую колбу вместимостью 100 см3, добавляют 2 г борной кислоты и 50 см3 соляной кислоты. Умеренно кипятят на плитке в течение 30 мин. Раствор охлаждают, переводят вместе с осадком в мерную колбу вместимостью 250 см3, доливают до метки водой и перемешивают. Часть раствора фильтруют через сухой, плотный фильтр в сухую колбу вместимостью 100 см3. Первые порции фильтрата отбрасывают. Отбирают 10 см3 анализируемого раствора в мерную колбу вместимостью 50 см3, добавляют 14 см3 смешанного реактива, обмывают горло колбы тонкой струей воды и оставляют на 15 мин.
Затем раствор в колбе доливают до метки водой, перемешивают и сразу измеряют оптическую плотность раствора, применяя светофильтр с максимумом светопропускания 597 нм в кювете с оптимальной толщиной поглощающего свет слоя раствора 50 мм.
Раствором сравнения служит раствор контрольного опыта, проведенный через все стадии анализа.
По величине оптической плотности испытуемого раствора находят массовую долю фосфора по градуировочному графику.
12.1.3.2 Для построения градуировочного графика в десять мерных колб вместимостью 50 см3 отмеривают микробюреткой 1, 2, 3, 4, 5, 6, 7, 8, 9 и 10 см3 стандартного раствора Б. В одиннадцатую колбу раствор не отмеривают.
В каждую колбу приливают по 14 см3 смешанного реактива и далее поступают, как указано в 12.1.3.1.
Раствором сравнения служит раствор, не содержащий стандартный раствор фосфора.
По полученным значениям оптической плотности растворов и известным содержаниям фосфора строят градуировочный график.
Правильность построения градуировочного графика проверяют по стандартному образцу состава флюоритового концентрата.
12.1.4 Обработка результатов
12.1.4.1 Массовую долю фосфора И/р, %, вычисляют по формуле
ы/ Л7«У-100
(12.1)
Wp =—!,
н Ц-т-1000
где т1 — масса фосфора, найденная по градуировочному графику, мг;
V — объем испытуемого раствора в мерной колбе, см3;
— объем аликвотной части испытуемого раствора, см3;
т — масса навески плавикового шпата, г.
12.1.4.2 Предел повторяемости г и предел воспроизводимости R анализа при доверительной вероятности Р= 0,95 не должны превышать допускаемых расхождений, приведенных в таблице 18.
Таблица 18
Массовая доля фосфора, % | Допускаемые расхождения, % | |||||
предел повторяемости г | предел воспроизводимости R | |||||
От | 0,010 | ДО | 0,050 | включ. | 0,004 | 0,005 |
Св. | 0,050 | » | 0,100 | » | 0,007 | 0,010 |
» | 0,100 | » | 0,300 | » | 0,012 | 0,015 |
» | 0,300 | » | 0,500 | » | 0,015 | 0,020 |
12.2 Молибдофосфатный фотометрический метод определения содержания фосфора
12.2.1 Назначение методаНастоящий подраздел устанавливает молибдофосфатный фотометрический метод определения общего содержания фосфора в плавиковом шпате для производства плавиковой кислоты.
Метод распространяется на материалы с общим содержанием фосфора (в пересчете на фосфат-ион РО43-) в пределах 0,02 %—1,0 % (по массе).
12.2.2 Сущность метода
Удаление диоксида кремния обработкой навески вначале плавиковой, а затем серной кислотами. Растворение остатка и приготовление раствора для испытаний. Образование молибдофосфатного комплекса желтого цвета и фотометрическое измерение абсорбции комплекса после экстракции смесью н-бутанола и хлороформа при длине волны 330 нм.
12.2.3 Реактивы
В процессе выполнения анализа следует использовать реактивы только аналитической степени чистоты и только дистиллированную воду и воду эквивалентной чистоты.
Смесь растворителей, н-бутанол/хлороформ. Смешивают равные объемы этих двух растворителей.
Примечание — Эти реактивы при их вдыхании оказывают токсичное действие. Следует избегать попадание на кожу и в глаза.
Азотная кислота с плотностью 1,40 г/см3, раствор 68 % (по массе).
Азотная кислота, раствор 315 г/дм3.
Серная кислота с плотностью 1,84 г/см3, 96 %-ный раствор (по массе).
Плавиковая кислота, с плотностью 1,14 г/см3, 40 %-ный раствор (по массе).
Примечание — Плавиковая кислота оказывает чрезвычайно токсичное действие при вдыхании, при контакте с кожей, вызывает сильные ожоги. Сосуд следует хранить плотно закрытым в хорошо проветриваемом помещении. В случае попадания в глаза необходимо немедленно промыть большим количеством воды и обратиться к врачу. Работать с плавиковой кислотой следует в соответствующей защитной одежде и в перчатках. В случае аварии, а также при недомогании следует немедленно обратиться к врачу (при возможности показать ярлык реактива).
Молибдат натрия, дигидрат Na2MoO4 ■ 2Н2О, раствор 30 г/дм3.
Фосфор, стандартный раствор А, соответствующий содержанию ионов РО43- 0,100 г в 1 дм3. Небольшое количество дигидрофосфата калия КН2РО4 высушивают в печи при температуре (105 ± 2) °C в течение 2 ч. Затем дают остыть в эксикаторе. Взвешивают с точностью до 0,0002 г 0,1433 г высушенного дигидрофосфата калия и количественно переносят в мерную колбу вместимостью 1000 см3. Растворяют в воде, разбавляют до метки и перемешивают. 1 см3 этого стандартного раствора содержит 100 мкг РО43-.
Фосфор, стандартный раствор Б, соответствующий содержанию ионов РО43- 0,010 г в 1 дм3. 100 см3 стандартного раствора А помещают в мерную колбу вместимостью 1000 см3, разбавляют до метки и перемешивают. 1 см3 такого стандартного раствора содержит 10 мкг РО43-.
12.2.4 Оборудование
Для проведения анализа применяют:
электропечь с устройством, позволяющим регулировать температуру (105 ± 2) °C;
платиновую чашку с диаметром приблизительно 90 мм;
спектрофотометр или фотометр, снабженный светофильтрами, обеспечивающими максимум 1 пропускание при длине волны приблизительно 330 нм;
оптические кюветы, изготовленные из кварца, с длиной оптического пути 14 или 50 мм.
12.2.5 Проба для анализа
В качестве пробы для анализа используют остаток после определения потери массы при 105 °C.
12.2.6 Проведение анализа
12.2.6.1 Навеска
Несколько граммов пробы для анализа по 12.2.6 истирают в агатовой ступке до прохождения через сито с размером отверстий 63 мкм. Истертый материал сушат 2 ч в печи при (105 ± 2) °C, дают остыть в эксикаторе и отвешивают с точностью до 0,0002 г примерно 0,1 г навески в платиновую чашку.
12.2.6.2 Контрольный опыт
Контрольный опыт следует проводить одновременно с анализом, следуя той же методике и используя те же количества всех реактивов, как и при анализе, но без навески.
12.2.6.3 Градуировочные графики подготавливают для двух пределов массовой доли фосфора (выраженного как РО43-):
- от 0,02 % до 0,2 % (график А);
- от 0,2 % до 1,0 % (график В).
а) Подготовка стандартных растворов для колориметрии
В серию мерных колб вместимостью 500 см3 помещают указанные в таблице 19 объемы стандартного раствора фосфора.
Таблица 19
Для градуировочного графика А | Для градуировочного графика В | ||
Стандартный раствор фосфора Б, см3 | Соответствующая масса РО43-, мкг | Стандартный раствор фосфора А, см3 | Соответствующая масса РО43", мкг |
о1) | 0 | 0 | 0 |
2,0 | 20,0 | 2,0 | 200,0 |
5,0 | 50,0 | 4,0 | 400,0 |
10,0 | 100,0 | 6,0 | 600,0 |
15,0 | 150,0 | 8,0 | 800,0 |
20,0 | 200,0 | 10,0 | 1000,0 |
1) Контрольный опыт на реактивы для градуировки. | |||
Каждый из этих растворов обрабатывают следующим образом: разбавляют водой до объема примерно 200 см3, добавляют 50 см3 раствора кислоты, 50 см3 раствора молибдата натрия разбавляют водой до метки и перемешивают. Аликвоту полученного раствора объемом 100 см3 переносят в делительную воронку на 200 см3, добавляют 20 см3 смеси растворителей и встряхивают в течение приблизительно 1 мин. После разделения фаз нижний слой отфильтровывают через фильтровальную бумагу в мерную колбу вместимостью 50 см3. Экстракцию и фильтрование повторяют с еще одной порцией смеси растворителей объемом 20 см3, после чего фильтровальную бумагу промывают несколькими кубическими сантиметрами смеси растворителей, собирая фильтрат в ту же колбу. Содержимое колбы разбавляют до метки смесью растворителей и перемешивают.
б) Фотометрические измерения
Измерения проводят либо с помощью спектрофотометра при длине волны 330 нм, либо с помощью фотометра с соответствующими светофильтрами после настройки прибора на ноль абсорбции относительно смеси растворителей. Для подготовки градуировочного графика А следует использовать кюветы с толщиной оптического слоя 40 или 50 мм, а для подготовки градуировочного графика В — кюветы с толщиной оптического слоя 10 мм. Из измеренных значений абсорбции стандартных растворов по данному подпункту вычитают измеренное значение абсорбции раствора контрольного опыта.
в) Построение градуировочных графиков
Градуировочный график строят для каждого из двух диапазонов содержания фосфора в пробе для испытаний, откладывая на оси абсцисс массы РО43- в микрограммах, содержащиеся в 500 см3 для каждого из стандартных растворов по данному подпункту, а по оси ординат — соответствующие величины абсорбции с учетом поправки на абсорбции раствора контрольного опыта.
12.2.6.4 Определение
а) Подготовка раствора для анализа
Пробу для анализа (см. 12.2.6.1) обрабатывают в платиновой чашке следующим образом: добавляют 10 см3 раствора плавиковой кислоты, осторожно выпаривают досуха на плитке в вытяжном шкафу с хорошей тягой и дают остыть. Эту операцию повторяют с еще одной порцией раствора плавиковой кислоты объемом 10 см3. Затем добавляют 10 см3 раствора серной кислоты и выпаривают до тех пор, пока содержимое чашки не станет лишь слегка влажным, дают остыть.
При необходимости удаляют любые органические примеси (признаком которых является коричневое окрашивание) добавлением нескольких капель раствора азотной кислоты и нагреванием. Содержимому снова дают остыть.
Далее добавляют 50 см3 раствора азотной кислоты и 50 см3 воды, осторожно нагревают в течение нескольких минут и количественно переносят содержимое платиновой чашки в стакан вместимостью 400 см3. Стакан нагревают и медленно кипятят в течение примерно 30 мин для растворения остатка. Содержимому дают остыть и количественно переносят в мерную колбу вместимостью 500 см3, разбавляют примерно 200 см3 воды и затем добавляют 50 см3 раствора молибдата натрия. Доводят до метки водой и перемешивают. Дают осесть осадку или отделяют его с помощью центрифуги.
Аликвоту этого раствора объемом 100 см3 переносят в делительную воронку вместимостью 200 см3 и, применяя смесь растворителей, проводят экстракцию по методике, описанной в 12.2.6.3.
б) Фотометрические измерения
Используя кюветы с толщиной оптического слоя 40 или 50 мм для растворов с массовой долей РО43- в диапазоне 0,02 %—0,2 % и кюветы с толщиной оптического слоя 10 мм для раствора с массовой долей в диапазоне 0,2 %—1,0 %, проводят фотометрическое измерение экстрактов, полученных из испытуемого раствора по данному подпункту, и экстракта из раствора контрольного опыта по 12.2.6.2, предварительно установив прибор на ноль абсорбции относительно смеси растворителей.
12.2.7 Обработка результатов
С помощью соответствующего градуировочного графика определяют массы РО43- в испытуемом растворе и в растворе контрольного опыта в соответствии с измеренными величинами абсорбции.
Общую массовую долю фосфора 1/1фо43-> %, вычисляют по формуле
100
(12.2)
Р°?~ ю6 ’
где т0 — масса навески (12.2.6.1), г;
т1 — масса РО43-, содержащаяся согласно результатам анализа в 500 см3 испытуемого раствора (12.2.6.4), мкг;
т2 — масса РО43-, содержащаяся согласно результатам анализа в 500 см3 раствора контрольного опыта (12.2.6.2), мкг.
12.2.8 Отчет об анализе
Отчет об анализе должен содержать:
- идентификацию пробы;
- ссылку на использованный метод;
- результаты и способ их выражения;
- любые особенности, отмеченные во время анализа;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
13 Методы определения содержания оксида магния
13.1 Фотоколориметрический метод (при массовой доле оксида магния от 0,02 % до 3 %)
13.1.1 Сущность методаНастоящий раздел устанавливает фотоколориметрический метод определения массовой доли оксида магния.
Метод основан на абсорбции гидроксидом магния титанового желтого в щелочной среде (pH 12) и фотометрировании окрашенного раствора. Мешающее влияние железа, марганца и алюминия устраняют связыванием их триэтаноламином в растворимые бесцветные комплексные соединения.
13.1.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
фотоэлектроколориметр;
кислоту азотную по ГОСТ 4461, разбавленную 1:1;
кислоту соляную по ГОСТ 3118, разбавленную 1:1, 1:5 и 1:99;
спирт этиловый (гидролизный), ректификованный;
триэтаноламин, разбавленный 1:3;
хлорид кальция, не содержащий магний;
желатин пищевой по ГОСТ 11293, свежеприготовленный раствор с массовой долей 1 %; готовят следующим образом: 1 г желатина помещают в стакан вместимостью 300 см3, приливают 30—40 см3 воды и оставляют на 1 ч, периодически перемешивая. Затем стакан с содержимым помещают в нагретую до кипения воду, перемешивают до растворения, доливают водой до 100 см3 и вновь перемешивают;
спирт поливиниловый, раствор с массовой долей 1 %, готовят в день применения, растворяя навеску при кипении в течение 5 мин;
гидроксид натрия по ГОСТ 4328, раствор 1 моль/дм3; готовят следующим образом: 40 г гидроксида натрия растворяют в воде в мерной колбе вместимостью 1000 см3, доливают до метки водой и перемешивают. Раствор выдерживают в течение 2 сут и хранят в полиэтиленовом сосуде, плотно закрыв пробкой;
смесь карбонатов калия и натрия по ГОСТ 4332;
титановый желтый (аммонийная соль), раствор с массовой долей 0,5 % в растворе этилового спирта с массовой долей 50 % и раствор с массовой долей 0,01 %, приготовленный следующим образом: к 2 см3 раствора с массовой долей 0,5 % приливают 2,5 см3 раствора поливинилового спирта, доливают водой до объема 100 см3 и перемешивают. Растворы хранят в склянках из темного стекла;
магний металлический, не ниже 99,95 %. Для удаления оксида с поверхности магний промывают соляной кислотой, разбавленной 1:5, затем три — пять раз водой и высушивают при 105 °C — 110 °C;
стандартные растворы оксида магния:
раствор А, приготовленный следующим образом: 0,6032 г металлического магния растворяют в 25 см3 соляной кислоты, разбавленной 1:1, в мерной колбе вместимостью 1000 см3, доливают водой до метки и перемешивают. 1 см3 раствора А соответствует 1 мг оксида магния; раствор Б, приготовленный следующим образом: отбирают 100 см3 раствора А в мерную колбу вместимостью 1000 см3, прибавляют 2,96 г хлорида кальция, доливают до метки водой и перемешивают. 1 см3 раствора Б соответствует 0,1 мг оксида магния.
13.1.3 Проведение анализа
13.1.3.1 Навеску пробы плавикового шпата массой 0,25—0,5 г (в зависимости от массовой доли оксида магния) помещают в коническую колбу вместимостью 250 см3, добавляют 30 см3 азотной кислоты, разбавленной 1:1, и далее содержимое колбы кипятят в течение 30 мин, покрыв часовым стеклом. Далее раствор выпаривают досуха, приливают 10 см3 концентрированной азотной кислоты и снова выпаривают досуха. Выпаривание с азотной кислотой повторяют. Для удаления окислов азота приливают 10 см3 соляной кислоты и вновь выпаривают досуха. Выпаривание с соляной кислотой повторяют дважды. Сухой остаток смачивают 7 см3 соляной кислоты, приливают 20 см3 горячей воды и кипятят 2—3 мин, покрыв часовым стеклом, до растворения растворимых солей. Нерастворимый остаток отфильтровывают на плотный фильтр, промывают пять-шесть раз горячей водой, подкисленной соляной кислотой, собирая фильтрат и промывные воды в стакан вместимостью 300 см3.
При массовой доле в плавиковом шпате минерала слюды, содержащей магний, нерастворимый остаток доплавляют. Для этого фильтр с нерастворимым остатком помещают в платиновый тигель, подсушивают, озоляют и прокаливают в муфельной печи при 600 °C — 700 °C в течение 30 мин.
Содержимое тигля охлаждают, прибавляют пяти-, шестикратное количество смеси карбонатов калия и натрия по отношению к осадку, перемешивают и сплавляют в муфельной печи при 850 °C — 900 °C до однородной массы.
Плав охлаждают, помещают вместе с тиглем в стакан с первоначальным фильтратом, выщелачивают, тигель извлекают из стакана и обмывают водой.
Раствор в стакане выпаривают до получения влажных солей. Соли растворяют в 5 см3 соляной кислоты, приливают пипеткой 10 см3 раствора желатина, 20 см3 горячей воды, перемешивают в течение 5 мин и фильтруют раствор в мерную колбу вместимостью 200 см3. Осадок на фильтре промывают восемь — десять раз горячей соляной кислотой, разбавленной 1:99. Раствор в колбе доливают водой до метки и перемешивают. Отбирают аликвотную часть раствора 2—10 см3 (в зависимости от массовой доли оксида магния) в мерную колбу вместимостью 50 см3, приливают 3 см3 разбавленного триэтаноламина, 5 см3 0,01 %-ного раствора титанового желтого, 5 см3 раствора поливинилового спирта, перемешивают и приливают из бюретки по каплям раствор гидроксида натрия до изменения окраски жидкости из лимонно-желтой в желто-розовую и в избыток 5 см3. Раствор доливают до метки колбы водой и перемешивают. Через 15 мин измеряют оптическую плотность раствора на фотоэлектроколориметре, применяя светофильтр с максимумом пропускания 545 нм, в кювете с толщиной поглощающего свет слоя раствора 50 мм.
Раствором сравнения служит раствор контрольного опыта с добавлением хлорида кальция в количестве, соответствующем массовой доле его в пробе.
По величине оптической плотности анализируемого раствора устанавливают массовую долю оксида магния по градуировочному графику.
13.1.3.2 Для построения градуировочного графика в мерные колбы вместимостью по 50 см3 микробюреткой отмеривают 0,05; 0,1; 0,2; 0,4; 0,6; 0,8; 1,0 см3 стандартного раствора Б, что соответствует 0,005; 0,01; 0,02; 0,04; 0,06; 0,08; 0,1 мг оксида магния. Во все колбы приливают воды до объема раствора 10 см3, по 3 см3 разбавленного триэтаноламина, по 5 см3 раствора титанового желтого с массовой долей 0,01 %, по 5 см3 раствора поливинилового спирта, перемешивают и далее анализ продолжают по 13.1.3.1.
Для построения градуировочного графика берут среднее арифметическое результатов трех измерений оптической плотности каждого раствора.
По полученным средним значениям оптической плотности растворов и известным содержаниям оксида магния строят градуировочный график.
13.1.4 Обработка результатов
13.1.4.1 Массовую долю оксида магния И/Мд0, %, вычисляют по формуле
m,V-100
^-Ц.т-ЮОО’ <13-1>
где т — масса навески пробы, г;
т1 — масса оксида магния, найденная по градуировочному графику, мг;
V — объем анализируемого раствора, см3;
V1 — объем аликвотной части раствора, см3.
13.1.4.2 Разность результатов пределов повторяемости при доверительной вероятности Р = 0,95 не должна превышать допускаемых расхождений, приведенных в таблице 20.
Таблица 20
Массовая доля оксида магния, % | Допускаемое расхождение d, % | ||||
До | 0,05 | включ. | 0,006 | ||
Св. | 0,05 | » | 0,1 | » | 0,0065 |
» | 0,1 | » | 0,3 | » | 0,03 |
» | 0,3 | » | 1 | » | 0,06 |
» | 1 | » | 3 | » | 0,14 |
Если расхождение между результатами двух пределов повторяемости превышает приведенную величину, определение повторяют.
За окончательный результат анализа принимают среднее арифметическое результатов двух пределов повторяемости.
13.2 Атомно-абсорбционный метод (при массовой доле оксида магния от 0,005 % до 3 %)
13.2.1 Сущность методаНастоящий подраздел устанавливает атомно-абсорбционный метод определения массовой доли оксида магния.
Метод основан на измерении оптической плотности при фотометрии пламени с помощью фотометра, которая пропорциональна концентрации определяемого элемента в анализируемом растворе, вводимого в пламя газовой горелки. Пламя бутан-пропан-воздушное.
13.2.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
фотометр типа «Спектр-1»;
реактивы и растворы, приведенные в 13.1.2;
стандартный раствор оксида магния, приготовленный следующим образом: отбирают пипеткой 10 см3 стандартного раствора А, приготовленного по 13.1.2, в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают. 1 см3 стандартного раствора соответствует 0,01 мг оксида магния;
хлорид кальция, не содержащий магний, раствор, приготовленный следующим образом: 2,96 г соли помещают в мерную колбу вместимостью 1000 см3, доливают водой до метки и перемешивают.
13.2.3 Проведение анализа
13.2.3.1 При массовой доле оксида магния в плавиковом шпате до 0,04 % раствор, приготовленный по 13.1.3.1 в мерной колбе вместимостью 200 см3, фотометрируют при аналитической линии магния 285,2 нм. Сначала в пламя горелки вводят раствор контрольного опыта с добавлением хлорида кальция в количестве, соответствующем массовой доле его в пробе. Затем в пламя горелки вводят анализируемый раствор.
По оптической плотности пламени анализируемого раствора с учетом оптической плотности пламени раствора контрольного опыта устанавливают концентрацию оксида магния по градуировочному графику.
Фотометрирование для каждой навески пробы проводят три раза и вычисляют среднюю арифметическую величину концентрации оксида магния.
13.2.3.2 При массовой доле оксида магния в плавиковом шпате свыше 0,04 % от раствора, приготовленного по 13.1.3.1 в мерной колбе вместимостью 200 см3, отбирают аликвотную часть 1—10 см3 (в зависимости от массовой доли оксида магния) в мерную колбу вместимостью 100 см3, доливают до метки водой и фотометрируют, как указано в 13.2.3.1.
13.2.3.3 Для построения градуировочного графика в мерные колбы вместимостью по 50 см3 отбирают 0,5; 1,0; 2,5; 5,0 см3 стандартного раствора оксида магния. В каждую колбу приливают раствор хлорида кальция до метки и фотометрируют. Концентрация приготовленных растворов соответствует 0,1; 0,2; 0,5 и 1 мкг/см3 оксида магния.
Для построения градуировочного графика вычисляют среднее арифметическое значение результатов трех измерений оптической плотности пламени для каждого раствора.
По полученным средним значениям оптической плотности растворов и известным концентрациям оксида магния строят градуировочный график.
13.2.4 Обработка результатов
13.2.4.1 Массовую долю оксида магния Х^, %, вычисляют по формуле где С — концентрация оксида магния, найденная по градуировочному графику, мкг/см3;
*1 =
С-У-Ц-100
У2-т-106
(13.2)
V — объем всего анализируемого раствора, см3;
— объем разбавления аликвотной части раствора, см3;
т — масса навески пробы, г;
V2 — объем аликвотной части раствора, см3.
13.2.4.2 Допускаемое расхождение между результатами двух пределов повторяемости приведено в п.13.1.4.2.
14 Метод определения содержания оксида стронция
14.1 Сущность метода
Настоящий раздел устанавливает эмиссионный пламеннофотометрический метод определения массовой доли стронция в пересчете на оксид стронция.
Метод основан на измерении интенсивности спектральной линии стронция в пламени.
14.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
фотометр пламенный эмиссионный с монохроматором;
кислоту соляную по ГОСТ 3118, разбавленную 1:1;
смесь карбоната калия и карбоната натрия безводную по ГОСТ 4332;
карбонат натрия безводный по ГОСТ 83, раствор с массовой долей 10 г/дм3;
карбонат кальция по ГОСТ 4530, ос. ч., раствор с массовой долей 125 г/дм3; готовят следующим образом: 125 г соли растворяют в воде в мерной колбе вместимостью 1000 см3, приливают 300 см3 разбавленной 1:1 соляной кислоты, доливают до метки водой и перемешивают. Раствор содержит 50 г/дм3 кальция;
карбонат стронция по ГОСТ 2821;
стандартные растворы стронция:
раствор А, приготовленный следующим образом: 1,427 г карбоната стронция растворяют в 100 см3 воды с добавлением 2—3 см3 разбавленной 1:1 соляной кислоты и кипятят 2—3 мин для удаления углекислого газа. Раствор охлаждают и переливают в мерную колбу вместимостью 1000 см3, доливают до метки водой и перемешивают. 1 см3 раствора А соответствует 1 мг оксида стронция;
раствор Б, приготовленный следующим образом: отбирают 100 см3 раствора А в мерную колбу вместимостью 1000 см3, доливают до метки прокипяченной и охлажденной водой и перемешивают. 1 см3 раствора Б соответствует 0,1 мг оксида стронция.
14.3 Проведение анализа
14.3.1 Навеску пробы плавикового шпата массой 1 г помещают в платиновый тигель, смешивают с 5 г смеси карбонатов калия и натрия и сплавляют при 850 °C — 900 °C в течение 15 мин.
Тигель с плавом охлаждают, помещают в стакан вместимостью 300 см3, приливают 150 см3 горячей воды и нагревают до полного выщелачивания. Тигель вынимают и обмывают водой над стаканом. Осадок отфильтровывают на плотный фильтр, промывают пять-шесть раз раствором карбоната натрия и пять-шесть раз горячей водой. Фильтрат отбрасывают. Осадок смывают с фильтра в стакан, где проводилось выщелачивание. Фильтр промывают 15 см3 теплой, разбавленной 1:1, соляной кислотой и 10 см3 этой же кислоты обмывают стенки стакана. Далее фильтр промывают семь-восемь раз горячей водой. Фильтрат и промывные воды собирают в стакан, где проводилось выщелачивание.
Раствор в стакане кипятят 5 мин для удаления углекислого газа, охлаждают, переливают в мерную колбу вместимостью 100 см3, доливают до метки водой и перемешивают.
Раствор фотометрируют при длине волны излучения 460,7 нм. Область спектра — видимая. Источником атомизации служит воздушно-бутан-пропановое или воздушно-ацетиленовое пламя.
По измеренной интенсивности спектральной линии стронция в пламени для анализируемого раствора устанавливают массовую долю оксида стронция по градуировочному графику.
14.3.2 Для построения градуировочного графика в мерные колбы вместимостью по 100 см3 отбирают 0,5; 1,0; 2,0; 5,0 и 10 см3 стандартного раствора Б. В каждую колбу приливают по 10 см3 раствора карбоната кальция, доливают до метки водой и перемешивают. Массовая доля приготовленных растворов соответствует 0,5; 1,0; 2,0; 5,0 и 10 мкг/см3 оксида стронция. Растворы фотометрируют, как указано в 14.3.1.
Далее вычисляют среднее арифметическое результатов трех измерений интенсивности спектральной линии стронция в пламени для каждого стандартного раствора.
По полученным средним значениям интенсивности спектральной линии стронция для стандартных растворов и известным массовым долям оксида стронция строят градуировочный график.
14.4 Обработка результатов
14.4.1 Массовую долю оксида стронция в плавиковом шпате H/Sr0, %, вычисляют по формуле
СУ-100
WStO =---—Q~>
(14.1)
л?-10°
где С — массовая доля оксида стронция, найденная по градуировочному графику, мкг/см3;
V — объем мерной колбы, см3;
т — масса навески пробы, г.
14.4.2 Разность результатов пределов повторяемости при доверительной вероятности Р = 0,95 не должна превышать допускаемых расхождений, приведенных в таблице 21.
Таблица 21
Массовая доля оксида стронция в плавиковом шпате, % | Допускаемое расхождение d, % | ||||
От | 0,005 | ДО | 0,01 | включ. | 0,0025 |
Св. | 0,01 | » | 0,03 | » | 0,003 |
» | 0,03 | » | 0,06 | » | 0,006 |
» | 0,06 | » | 0,15 | » | 0,01 |
Если расхождение между результатами двух пределов повторяемости превышает приведенное значение, определение повторяют.
За окончательный результат анализа принимают среднее арифметическое результатов двух пределов повторяемости.
14.4.3 Контроль точности измерений проводят в соответствии с разделом 3.
15 Метод определения содержания оксида бария
15.1 Сущность метода
Настоящий раздел устанавливает спектрографический метод определения массовой доли бария в пересчете на оксид бария.
Метод основан на нахождении спектральной линии искомого элемента в пробе при испарении ее в смеси с угольным порошком из канала угольного электрода в дуге переменного тока и измерении ее интенсивности.
Элементом сравнения служит кальций.
15.2 Реактивы, растворы и оборудование для проведения испытания
Для проведения анализа применяют:
спектрограф дифракционный марок ДФС-13, ДФС-8 или спектрограф марки ИСП-51 с камерой УФ-85 или аналогичный с разрешающей способностью (линейной дисперсией) 0,3—0,6 нм/мм;
источник питания дуги переменного тока;
микрофотометр типа МФ-2;
станок и фрезы для заточки электродов;
штатив вертикальный с осветителем;
ступку агатовую или яшмовую;
электроды угольные спектральные марки С-2 или С-3 диаметром 6 мм;
фотопластинки «Микро» и спектральные типов I и II чувствительностью соответственно 60— 130 ед., 1—2 ед. и 16 ед., размером 9x12 см;
порошок угольный, истертый до крупности 0,063 мм;
сульфат бария по ГОСТ 3158;
фторид кальция;
спирт этиловый (гидролизный) ректификованный;
гидрохинон (парадиоксибензол) по ГОСТ 19627;
бромид калия по ГОСТ 4160;
метол (пара-метиламинофенолсульфат) по ГОСТ 25664;
карбонат натрия безводный по ГОСТ 83;
сульфит натрия безводный по ГОСТ 195;
тиосульфат натрия по ГОСТ 27068, фиксаж, раствор с массовой долей 250 г/дм3.
15.3 Подготовка к проведению испытания
15.3.1 Электроды для анализа готовят следующим образом:
верхний угольный электрод затачивают на усеченный конус на длину 5 мм с площадкой диаметром 2 мм. В нижнем угольном электроде высверливают кратер диаметром 4 мм, глубиной 3 мм и толщиной стенок 0,75 мм. Канал электрода должен иметь боковое отверстие диаметром 1 мм на глубине 2,8 мм.
15.3.2 Проявитель метоловый готовят следующим образом:
1 г метола растворяют в 500—700 см3 воды, прибавляют 12 г сульфита натрия, 5 г гидрохинона, 12 г карбоната натрия, 0,4 г бромида калия и приливают воды до 1000 см3.
15.3.3 В качестве стандартных образцов используют смеси, для приготовления которых все реактивы должны быть истерты до крупности 0,063 мм.
В качестве основы используют фторид кальция, в котором массовую долю бария определяют спектральным методом добавок. При этом барий определяют не менее чем из пяти отдельных навесок (по три параллельных экспозиции) и полученные результаты усредняют.
Стандартный образец готовят следующим образом: 1,5229 г сульфата бария и 8,4771 г фторида кальция тщательно истирают с добавлением спирта в ступке в течение 2 ч. Стандартный образец содержит 10 % оксида бария.
Далее отвешивают 1 г стандартного образца и 9 г основы и полученную смесь тщательно истирают с добавлением спирта. 10 г этого стандартного образца содержат 1% оксида бария.
Стандартные образцы, содержащие 0,001 %; 0,003 %; 0,005 %; 0,01 %; 0,03 %; 0,05 %; 0,01 %; 0,3 % и 0,5 % оксида бария, готовят последовательным разбавлением предыдущего стандартного образца основой. Перемешивание производят тем же способом, что и при приготовлении исходного стандартного образца.
Массовую долю оксида бария в стандартных образцах рассчитывают как сумму введенной массовой доли и массовой доли его в основе.
Стандартные образцы смешивают с угольным порошком в соотношении 1:1 по массе.
15.4 Проведение анализа
15.4.1 Навеску плавикового шпата массой 0,1 г тщательно истирают в ступке с 0,1 г угольного порошка с добавлением спирта. Полученной смесью наполняют по объему отверстия трех угольных электродов и сжигают в дуге переменного тока при силе тока разряда 12 А. Межэлектродный промежуток 1,5 мм. Спектр экспонируют 1,5 мин. Система освещения входной щели спектрографа — трехлинзовая. Ширина щели 0,01 мм. Расстояние между электродами поддерживают постоянным в течение всего времени экспонирования, наблюдая проекции электродов на промежуточной диафрагме высотой 5 мм.
Каждую навеску и стандартный образец сжигают по два раза на одну и ту же фотопластинку. Продолжительность проявления фотопластинок указана на их упаковке.
На спектрограмме с помощью микрофотометра измеряют почернение аналитической линии определяемого элемента и элемента сравнения. Ступень ослабителя выбирают в зависимости от интенсивности линии бария и линии сравнения.
Аналитические линии и интервалы определяемых массовых долей оксида бария указаны в таблице 22.
Таблица 22
Аналитические линии,нм | Интервал определяемых массовых долей, % |
Ва 455.4 Са 326,9 Ва 307,1 Са 451,2 | 0,001—0,020 0,01—1,00 |
В спектрах стандартных образцов и анализируемой навески плавикового шпата находят разность почернений AS между аналитической линией и линией сравнения.
15.4.2 По данным фотометрирования стандартных образцов строят градуировочный график в координатах AS—IgC, где С — массовая доля оксида бария в стандартных образцах.
15.5 Обработка результатов
15.5.1 По данным измерения почернения аналитической линии бария в спектре анализируемой навески плавикового шпата по градуировочному графику находят массовую долю оксида бария в пробе.
15.5.2 Разность между результатами предела повторяемости и результатами анализа при доверительной вероятности Р = 0,95 не должна превышать допускаемых расхождений, приведенных в таблице 23.
Таблица 23
Массовая доля оксида бария в плавиковом шпате, % | Допускаемое расхождение d, % | ||||
От | 0,001 | до | 0,005 | включ. | 0,0005 |
Св. | 0,005 | » | 0,01 | » | 0,0025 |
» | 0,01 | » | 0,03 | » | 0,003 |
» | 0,03 | » | 0,06 | » | 0,008 |
» | 0,06 | » | 0,10 | » | 0,01 |
» | 0,10 | » | 0,30 | » | 0,02 |
» | 0,30 | » | 1,00 | » | 0,03 |
Если расхождение между результатами двух пределов повторяемости превышает приведенное значение, определение повторяют.
За окончательный результат анализа принимают среднее арифметическое результатов двух пределов повторяемости.
15.5.3 Контроль точности измерений проводят в соответствии с разделом 3.
16 Метод определения флотационных реагентов
16.1 Назначение метода
Настоящий раздел устанавливает гравиметрический метод определения флотационных реагентов в плавиковом шпате, используемом для производства плавиковой кислоты. Метод распространяется на продукты, подвергавшиеся флотационной обработке, с массовой долей флотационных реагентов, равной или более 0,002 % в сухом материале.
16.2 Сущность метода
Обработка навески смесью разбавленной соляной кислоты и органического растворителя. Удаление нерастворимого плавикового шпата путем фильтрования под вакуумом. Отделение органической фазы, содержащей флотационный реагент, выпаривание растворителя и взвешивание осадка.
16.3 Реактивы
В ходе анализа используют реактивы только аналитической степени чистоты и только дистиллированную воду или воду эквивалентной чистоты.
Кислота соляная плотностью 1,19 г/см3, раствор с массовой долей 38 %.
Растворитель: дважды перегнанный трихлортрифторэтан-1,1,2 или диэтиловый эфир.
16.4 Оборудование
Для проведения анализа применяют:
прибор для фильтрования под вакуумом, состоящий из воронки Бюхнера диаметром 120 мм и с соответствующей фильтровальной бумагой и колбы Бюхнера вместимостью 1000 см3;
делительную воронку вместимостью 1000 см3;
механическую мешалку с лопастью диаметром 400 мм, приводимую в движение электромотором.
16.5 Проведение анализа
16.5.1 НавескаВ химический стакан вместимостью 1000 см3 отвешивают с точностью до 0,1 г 500 г лабораторной пробы, высушенной в соответствии с разделом 4.
16.5.2 Определение
В химический стакан, содержащий навеску, добавляют 300 см3 воды, 20 см3 раствора соляной кислоты и 200 см3 растворителя. Энергично перемешивают с помощью мешалки в течение 30 мин. Фильтруют на приборе для фильтрования под вакуумом, промывают осадок растворителем общим объемом 100 см3, добавляемым небольшими порциями. Переводят фильтрат, который содержит две фазы, из колбы Бюхнера в делительную воронку и обмывают колбу небольшим количеством растворителя. Отделяют нижнюю фазу, пропуская жидкость через фильтровальную бумагу, для того чтобы удалить воду, содержащуюся в растворителе, и собирают эту фазу в плоский фарфоровый сосуд. Фарфоровый сосуд помещают на паровую баню и выпаривают растворитель под вытяжкой в целях доведения объема до нескольких кубических сантиметров. Осадок количественно переносят в химический стакан вместимостью примерно 50 см3, предварительно высушенный при температуре примерно 100 °C, охлажденный в эксикаторе и взвешенный с точностью до 0,001 г. Тщательно обмывают растворителем сосуд, помещают в химический стакан на паровую баню, выпаривают досуха, охлаждают в эксикаторе и взвешивают с точностью до 0,001 г.
16.6 Обработка результатов
Массовую долю флотационных реагентов Х3, выраженную в процентах по массе высушенного продукта, вычисляют по формуле
ль-100
Х3=-3—, 16.1
/«о
где т1 — масса осадка, г;
т0 — масса навески, г.
16.7 Отчет об анализе
Отчет об анализе должен включать:
- ссылку на используемый метод;
- результаты и способ их выражения;
- любые особенности, замеченные во время определения;
- операции, не предусмотренные настоящим стандартом или стандартом, на который приведена ссылка, или рассматриваемые как необязательные.
17 Метод определения гранулометрического состава
17.1 Сущность метода
Настоящий раздел устанавливает гравиметрический метод определения гранулометрического состава ситовым анализом.
Сущность метода заключается в определении количественного распределения материала по крупности путем рассева на одном или нескольких ситах с последующим весовым определением полученных классов крупности и вычислении их выхода в процентах от общей массы пробы, взятой для ситового анализа.
17.2 Оборудование для проведения испытания
Для проведения анализа применяют:
сетки с квадратными отверстиями по ГОСТ 6613 и сетки с круглыми отверстиями по ГОСТ 3306; встряхиватели механические;
грохоты лабораторные механические с соответствующими сетками;
весы лабораторные с погрешностью взвешивания не более 0,5 % от массы взвешиваемого материала.
17.3 Метод отбора проб
17.3.1 Отбор общей пробы для определения гранулометрического состава — по ГОСТ 14180.
17.3.2 Количество разовых проб, отбираемых от партии плавикового шпата, определяют по таблице 1 ГОСТ 14180—80, при этом:
флотационный концентрат соответствует достаточно однородной руде;
гравитационный концентрат крупностью менее 5 мм — однородной;
гравитационный концентрат крупностью более 5 мм и окатыши — среднеоднородной;
кусковой плавиковый шпат — неоднородной.
17.3.3 Массу разовой пробы определяют по ГОСТ 14180. Масса разовой пробы должна быть не менее указанной в таблице 24.
Таблица 24
Размер максимального куска, мм | Масса разовой пробы, кг |
0,4 | 0,5 |
5,0 | 1,0 |
50,0 | 4,0 |
250,0 | 10,0 |
Масса общей пробы для определения гранулометрического состава определяется как сумма всех разовых проб, отобранных от партии. Минимальная масса общей пробы qmin, кг, должна быть не менее величины, вычисляемой по формуле
qmin = 0,02с/2 + 0,5с/, (17.1)
где d— размер максимальных кусков опробуемого материала, мм.
17.4 Подготовка к проведению испытания
17.4.1 Общую пробу тщательно перемешивают и методом сокращения выделяют три навески для проведения анализа.
17.4.2 Массу навески для рассева определяют в зависимости от крупности анализируемого материала в соответствии с таблицей 25.
Таблица 25
Размер максимального куска, мм | Масса навески, кг |
0,4 | 0,1 |
1,0 | 0,25 |
3,0 | 1 |
5,0 | 2 |
30,0 | 5 |
50,0 | 20 |
150,0 | 100 |
250,0 | 350 |
17.5 Проведение анализа
17.5.1 Рассев навесок плавикового шпата крупностью менее 5 мм проводят на механическом встряхивателе, а навесок материала крупностью более 5 мм — на грохотах с механическим приводом.
Рассев материала крупностью менее 3 мм допускается производить вручную.
17.5.2 Выбор сит в каждом конкретном случае определяется техническими требованиями на тот или иной вид продукции, а также целью испытания.
Сита в наборе для рассева располагают в нисходящем порядке размеров отверстий начиная с самого крупного.
17.5.3 При рассеве навеску подают на сито либо порциями, либо непрерывным равномерным потоком, не допуская перегрузки сит. При этом необходимо следить, чтобы материал на нем располагался слоем толщиной, не превышающей двукратный размер максимального куска.
17.5.4 Время просеивания зависит от класса крупности и считается достаточным, если при дополнительном просеивании в течение 1 мин в подрешетный продукт выделится не более 0,1 % материала от массы взятой навески.
17.5.5 Каждый класс плавикового шпата, полученный в результате рассева, взвешивают и результаты записывают.
Потери материала при рассеве не должны превышать 2 % от массы взятой навески.
Величину потерь плавикового шпата прибавляют к самому мелкому классу крупности.
Если величина потерь плавикового шпата превышает допустимую, испытание повторяют.
17.6 Обработка результатов
17.6.1 Содержание остатка на каждом сите X, %, вычисляют по формуле
x = mr1°°t (17.2)
т
где т1 — масса остатка на сите данного класса крупности, кг;
т — масса навески, взятой для испытания, кг.
17.6.2 Окончательный результат анализа подсчитывают по следующей методике. Обозначают результаты испытаний первой, второй и третьей навесок (содержание класса крупности) соответственно Х1,Х2иХ3. _
17.6.3 Среднее арифметическое значение Х1 2 результатов испытаний первой и второй навесок вычисляют по формуле
17.6.4 Максимально допускаемое отклонение результатов испытаний первой и второй навесок
Е1 2 от среднего арифметического значения Х1 2 вычисляют по формуле
Ei2 = r-Xi2, (17.4)
где г — коэффициент, характеризующий максимально допускаемое отклонение результатов испытаний от среднего арифметического, числовое значение которого в зависимости от условий определения равно:
0,1 — при параллельном определении в одной лаборатории;
0,15 — при сравнительных определениях в разных лабораториях.
17.6.5 Расхождение между результатами испытаний двух навесок (Х1 - Х2) сравнивают с максимально допускаемым отклонением Е1 2.
Если (Х1 - Х2) Е1 2> то Х1 2 принимают за окончательный результат.
Если (Х1 - Х2) > Е1 2, то проводят испытание третьей навески и вычисляют среднее арифметическое результатов испытаний первой и третьей Х1 3, а также второй и третьей навесок Х2 3.
Соответственно вычисляют значение Е1 2 и Е2 3.
При сравнении расхождений результатов испытаний первой и третьей, а также второй и третьей навесок с максимально допускаемыми отклонениями могут быть следующие варианты:
если (Х1 - Х2) < Е1 3, и одновременно (Х2 - Х3) < Е2 3, то за окончательный результат определения принимают
*1, з + Хг, 3 = Х1 + Х2+2Х3. /И 7 сX
2 4 ’ ( '
если (Х1 - Х3) < Е1 3 и одновременно (Х2 - Х3) > Е2 3 или (Х1 - Х3) > Е1 3 и одновременно (Х2 - Х3) < Е2 3 то за окончательный результат определения принимают соответственно Х1 3илиХ2 3;
если (Х1 - Х3) > Е1 3 и одновременно (Х2 - Х3) > Е2 3, то определение повторяют на вновь отобранных навесках.
УДК 543.2:006.354 МКС 73.080
Ключевые слова: шпат плавиковый, метод определения влаги, метод определения карбоната кальция, метод определения фторида кальция, метод определения диоксида кремния, метод определения металлических оксидов Ме2О3, методы определения железа, метод определения серы (общей), методы определения серы (сульфидной), метод определения фосфора, метод определения содержания оксида магния, метод определения содержания оксида стронция, метод определения содержания оксида бария, метод определения флотационных реагентов, метод определения гранулометрического состава
Редактор Н.В. Таланова
Технический редактор В.Н. Прусакова
Корректор Е.Д. Дульнева
Компьютерная верстка И.Ю. Литовкиной
Сдано в набор 30.06.2023. Подписано в печать 05.07.2023. Формат 60х847в. Гарнитура Ариал.
Усл. печ. л. 6,05. Уч-изд. л. 5,47.
Подготовлено на основе электронной версии, предоставленной разработчиком стандарта
Создано в единичном исполнении в ФГБУ «Институт стандартизации» , 117418 Москва, Нахимовский пр-т, д. 31, к. 2.
1
В Российской Федерации действует ГОСТ Р 58144—2018.
В Российской Федерации действует СП 52.13330.2011 «СНиП 23-05-95 Естественное и искусственное освещение».
** В Российской Федерации действуют Правила устройства электроустановок (утверждены Министерством энергетики Российской Федерации Приказом от 8 июля 2002 г. № 204).
“* В Российской Федерации действуют Правила технической эксплуатации электроустановок потребителей (утверждены Министерством энергетики Российской Федерации Приказом от 13 января 2003 г. № 6).
*4 В Российской Федерации действуют Правила по охране труда при эксплуатации электроустановок (утверждены Министерством труда и социальной защиты Российской Федерации Приказом от 24 июля 2013 г. № 328н).
2
5 В Российской Федерации действует Порядок утверждения типа стандартных образцов или типа средств
3
измерений, внесения изменений в сведения о них (приложение № 2 к приказу Минпромторга России от 28 августа 2020 г. № 2905).
4
*6 В Российской Федерации действует СП 44.13330.2011 «СНиП 2.09.04-87* Административные и бытовые здания».

{kind=link}