ГОСТ Р 56893-2016/ISO/TS 17665-2:2009
Группа Р26
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
Стерилизация медицинской продукции
ВЛАЖНОЕ ТЕПЛО
Часть 2
Руководство по применению ИСО 17665-1
Sterilization of health care products. Moist heat. Part 2. Guidance on the application of ISO 17665-1
ОКС 11.080.01
ОКП 94 5120
Дата введения 2017-03-01
Предисловие
1 ПОДГОТОВЛЕН Обществом с ограниченной ответственностью "Фармстер" (ООО "Фармстер") на основе собственного аутентичного перевода на русский язык документа, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 383 "Стерилизация изделий медицинского назначения"
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 17 марта 2016 г. N 157-ст
4 Настоящий стандарт идентичен международному документу ISO/TS 17665-2:2009* "Стерилизация медицинской продукции. Влажное тепло. Часть 2. Руководство по применению ИСО 17665-1" (ISO/TS 17665-2:2009 "Sterilization of health care products - Moist heat - Part 2: Guidance on the application of ISO 17665-1").
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей. - .
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации, сведения о которых приведены в дополнительном приложении ДА
5 ВВЕДЕН ВПЕРВЫЕ
Правила применения настоящего стандарта установлены в ГОСТ Р 1.0-2012 (раздел 8). Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе "Национальные стандарты", а официальный текст изменений и поправок - в ежемесячном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
Введение
Правила, приводимые в настоящей технической спецификации, не предусматривают использования их в качестве контрольного чек-листа для оценки соответствия требованиям первой части стандарта ИСО 17665-1.
Данное руководство призвано оказать помощь в достижении одинакового понимания и внедрения требований ИСО 17665-1 путем предоставления разъяснений и приведения приемлемых методов достижения соответствия указанным требованиям. Документ освещает важнейшие аспекты и приводит примеры. Методы, отличающиеся от приведенных в данном руководстве, также могут использоваться. Однако при использовании альтернативных методов они должны иметь подтверждение своей эффективности в плане достижения соответствия со стандартом ИСО 17665-1.
Основная часть этого документа применима ко всем уставкам и параметрам в тех случаях, когда выполняется стерилизация влажным теплом. Приложения к данному руководству также детально оговаривают средства, обеспечивающие выполнение требований ИСО 17665-1 и представляющие лучшие современные практики.
Нумерация статей основной части данной технической спецификации соответствует нумерации статей стандарта ИСО 17665-1.
Медицинская продукция, обрабатываемая в учреждениях здравоохранения, включает в себя широкий ряд изделий с различными уровнями биологической нагрузки (загрязнений). Соответствующие и тщательно выполняемые процессы очистки и - при необходимости обеспечения безопасного обращения - деконтаминации крайне важны и обязательны для выполнения перед передачей продукции на стерилизацию.
Загрузки из разнородных изделий обычны для учреждений здравоохранения, при этом обрабатываемые объемы определяются как по историческим данным, так и по прогнозируемым запросам на стерильную продукцию. Учреждения здравоохранения обычно не оговаривают использование стерилизационных процессов, предназначенных для каких-либо отдельных медицинских изделий. В обычную практику учреждений здравоохранения также не входит определение биологической нагрузки конкретного медицинского изделия. Очень важно, чтобы определенные инструменты перед деконтаминацией разбирались и тщательно инспектировались после завершения процесса стерилизации. Необходимы также последующая сборка и оценка состояния. По этой причине инструкции изготовителя медицинского изделия (см. ИСО 17664) должны тщательно соблюдаться во всех аспектах очистки, дезинфекции, упаковки и стерилизации. Многие изделия могут быть полностью погружены в жидкость, и могут быть отмыты и дезинфицированы в автоматизированном оборудовании (см. ИСО 15883). Для изделий, которые не могут быть полностью погружены в жидкость или не выдерживают тепловую деконтаминацию, должны использоваться альтернативные способы дезинфекции, обеспечивающие дальнейшее безопасное обращение с этими изделиями. На местах должны иметься и использоваться такие процедуры и политики, которые гарантируют прохождение медицинского изделия через обработку, соответствующую его характеристикам. Особое внимание должно уделяться сушке и хранению стерильных медицинских изделий. Требования к упаковке медицинских изделий изложены в ИСО 11607-1 и ИСО 11607-2. Если многократные циклы стерилизации могут приводить к деградации и ограничивать срок службы медицинского изделия, изготовитель такого изделия должен указывать допустимое количество циклов его обработки.
При выборе медицинского изделия приоритет должны иметь такие его свойства, как легкость очистки и разборки.
Дополнительные указания, специфические для сферы здравоохранения, приводятся в приложении D к данной технической спецификации.
ИСО 17665 под общим названием "Стерилизация медицинской продукции. Влажное тепло" состоит из следующих частей:
Часть 1: Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий;
Часть 2: Руководство по применению ИСО 17665-1.
1 Область применения
Данная техническая спецификация обеспечивает общее руководство при разработке, валидации и текущем контроле процессов стерилизации влажным теплом и предназначена для разъяснения требований, изложенных в стандарте ИСО 17665-1. Указания, приведенные в настоящей технической спецификации, предназначены для продвижения правильных практик, имеющих отношение к процессам стерилизации влажным теплом и для оказания помощи тем, кто разрабатывает и валидирует процессы стерилизации влажным теплом в соответствии с требованиями стандарта ИСО 17665-1.
Примечание 1 - Структура основной части данной технической спецификации ИСО (статьи 1-12) соответствует структуре ИСО 17665-1, таким образом, указания и разъяснения, приведенные в отдельных статьях или параграфах данной части, относятся к требованиям соответствующей статьи или параграфа ИСО 17665-1. Например, указания относительно ИСО 17665-1, параграфа 5.2 приведены в параграфе 5.2 данного документа. Эти указания являются дополнением к указаниям, приведенным в ИСО 17665-1, приложении А. Смотрите также приложение Е.
Примечание 2 - Особые специфические соображения, характерные для процессов стерилизации, выполняемых в учреждениях здравоохранения, приведены в приложении D.
2 Нормативные ссылки
Перечисленные ниже нормативные документы (стандарты)* являются обязательными при применении настоящего стандарта. Для датированных ссылок используют только указанную в ссылке редакцию. Для недатированных ссылок используют последнюю редакцию документа, включая любые дополнения, изменения и поправки.
_______________
* Таблицу соответствия национальных стандартов международным см. по ссылке. - .
ИСО 17665-1:2006 Стерилизация медицинской продукции. Влажное тепло. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий (ISO 17665-1:2006, Sterilization of health care products - Moist heat - Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices).
Примечание - В нормативных ссылках ИСО 17665-1, приводятся опубликованные стандарты, содержание которых должно быть использовано для оказания помощи при демонстрации соответствия требованиям той статьи, в которой дается соответствующая ссылка. Некоторые из них нужны, в основном, для промышленной стерилизации влажным теплом или для изготовителей паровых стерилизаторов, и могут таким образом выходить за рамки типовой практики, используемой при выполнении стерилизации влажным теплом в учреждениях здравоохранения.
ИСО 17665-1 описывает ряд методов и процедур, которые могут быть использованы для мониторинга процессов стерилизации. Требующееся для этого оборудование обычно присутствует на рынке. Ряд нормативных ссылок описывает спецификации и методы испытаний, используемые коммерческими поставщиками для аттестации их продукции. Пользователи такой продукции должны быть уверены, что приобретенные ими продукты соответствуют требованиям цитируемых стандартов, но обычно у них нет необходимости изучать эти стандарты.
ИСО 17665-1 описывает использование упаковки, соответствующей требованиям ИСО 11607-1 и ИСО 11607-2. Учреждения здравоохранения должны приобретать такую упаковку, которая соответствует требованиям этих стандартов.
Один из методов валидации процессов, описанный в ИСО 17665-1, основан на установлении бионагрузки.
Стандарты серии ИСО 11737 оговаривают ряд микробиологических методов, используемых в этом процессе. Учреждения здравоохранения обычно не должны использовать данный подход к валидации процессов.
3 Термины и определения
В настоящей технической спецификации применены термины и определения, используемые в стандарте ИСО 17665-1, со следующими дополнениями:
3.1 испытания на стерильность (tests for sterility): Техническая операция, описанная в фармакопее, выполняемая на продукте, подвергнутом стерилизации
4 Элементы системы менеджмента качества
Применимы указания, приведенные в ИСО 17665-1:2006, приложение А.
Примечание - Особые соображения, относящиеся к учреждениям здравоохранения, приведены в D.2.
5 Описание стерилизующего агента
5.1 Стерилизующий агент
5.1.1 Влажное тепло - это вода при повышенных температурах. Влажное тепло может обеспечиваться путем подачи насыщенного пара или вырабатываться на месте за счет приложения тепловой энергии к воде, уже содержащейся в продукте. Влага выступает в роли среды, передающей тепловую энергию микроорганизмам.
5.1.2 Примеси, содержащиеся в стерилизующем агенте, могут быть как токсичными, так и вызывающими коррозию, и могут образовывать барьер между микроорганизмами и стерилизующим агентом. Они поступают вместе с водой, которая нагревается или превращается в пар, либо образуются в результате контакта между материалами и стерилизующим агентом в ходе выпаривания и подачи пара в стерилизатор (см. статьи 6, 7 и приложение А). Если уровень примесей в стерилизующем агенте может быть изменен за счет качества питающей воды, подаваемой в парогенерирующую систему, то характеристики питающей воды должны оговариваться.
5.2 Микробоцидная эффективность
Микробоцидная активность влажного тепла основана на температуре и продолжительности контакта между молекулами воды и микроорганизмами.
Для целей стерилизации влажным теплом существует ряд приемлемых комбинаций времени и температуры, приводимый в некоторых фармакопеях. В число этих комбинаций входят (но не ограничиваются только ими) и те, которые перечислены в таблице 1. Все перечисленные комбинации базируются на концепции "полной гибели" с фактором безопасности, который был установлен для насыщенного пара или воды, имеющих контакт с микроорганизмом. Перегретый пар ведет себя в большей степени подобно сухому воздуху (газу) и имеет низкую микробоцидную активность по сравнению с насыщенным паром.
Перегретый пар может образовываться в результате снижения давления и/или термодинамического сжатия насыщенного пара. Он может также образовываться в результате обезвоживания частей стерилизуемой загрузки, в особенности частей, содержащих натуральные волокна.
Условия образования перегретого пара могут быть минимизированы за счет применения правильных инженерных решений при конструировании систем подачи пара, например:
a) установка серии редукционных клапанов на трубопроводе подачи пара в стерилизационную камеру и обеспечение снижения давления в каждой секции в соотношении не более 2:1;
b) обеспечение скорости пара не более 25 м/сек;
c) обеспечение предварительной обработки (кондиционирования) материалов из натуральных волокон для достижения ими влажности более 40% перед стерилизацией.
Таблица 1 - Примеры комбинаций минимальных температур и времени, установленных для адекватных уровней микробной летальности в стерилизационных процессах
Температура, °С | Время, мин |
121 | 15 |
126 | 10 |
134 | 3 |
5.3 Воздействие на материалы
Воздействие на материалы в общем случае ограничивается деформацией, трещинами, разрывами, вызванными давлением и температурой стерилизующего агента.
5.4 Экологические факторы
Принципы систем экологического менеджмента могут быть применены и к процессам стерилизации влажным теплом. ИСО 14001 приводит спецификацию системы экологического менеджмента. ИСО 14040 приводит руководство по проектированию исследования оценки жизненного цикла. Должно рассматриваться присутствие вредных веществ в сливе стерилизатора. Дополнительные указания по этой статье даются в ИСО 14937:2009, приложение Е, подраздел Е.3.
6 Описание процесса и оборудования
Примечание - Целью этого действия является описание характеристик стерилизационного процесса в целом и описание характеристик оборудования, необходимого для выполнения стерилизационного процесса безопасным и воспроизводимым путем.
6.1 Процесс
6.1.1 Общие требования
Процесс стерилизации должен быть определен для каждого семейства продуктов и/или конфигураций загрузки, представляемой для стерилизации.
Параметры процесса должны быть применимы к используемому оборудованию. Они должны быть оптимизированы таким образом, чтобы для каждого семейства продуктов рутинно обеспечивались оговоренные условия экспозиции во всем объеме стерилизационной камеры, а максимальные температуры и скорости изменения переменных процесса (например, температуры и давления) не приводили к повреждениям или деградации продукта.
Спецификация процесса стерилизации должна включать в себя все параметры процесса, определяющие профиль экспозиции на протяжении всего рабочего цикла. Та часть рабочего цикла, в которой устанавливаются условия обеспечения летальности, должна быть идентифицирована, а также должны быть определены верхний и нижний пределы для каждого параметра процесса, способного повлиять как на летальность, так и на качество медицинского изделия.
Должны предприниматься меры для записи (регистрации) данных для оценки эффективности и приемлемости рутинного процесса стерилизации. Точность измерений должна определяться допусками, установленными для параметров процесса.
Если предлагается использовать существующий процесс стерилизации для обработки нового медицинского изделия, этот существующий процесс должен быть детализирован, и детализация должна содержать информацию и данные, достаточные для выполнения определения процесса (см. статью 8) для предлагаемого нового медицинского изделия (изделий) или новой конфигурации загрузки.
Нагрузка, идентифицированная для нового медицинского изделия или для новых условий загрузки, должна быть меньше или равна нагрузке, определенной для существующей стерилизационной загрузки (загрузок). Для некоторых семейств продуктов уверенность в том, что определенные условия экспозиции будут воспроизводимыми, может быть возможна только при условии, что размеры стерилизационной загрузки и ее конфигурация были четко определены.
Если должны использоваться биологические и химические индикаторы, они не могут заменить собой текущий мониторинг, измерение переменных процесса и любые иные периодические испытания/ проверки.
Совместимость нового медицинского изделия с наименее благоприятными условиями процесса стерилизации должна быть оценена. Такая оценка должна включать в себя допуски на параметры процесса, неопределенность (неточность) связанных с параметрами процесса измерений, а также качество питающих сред (см. приложение А).
Любые ограничения размеров и массы стерилизуемой загрузки и ее конфигурации должны быть определены и записаны в рабочие инструкции.
Соотношение между температурой, измеренной в контрольной точке, и температурой, измеренной в загрузке, должно быть определено для каждого семейства продуктов.
Качество медицинского изделия может зависеть от наличия примесей на его поверхности. Примеси и их максимально допустимая концентрация во всех жидкостях, входящих в контакт с медицинским изделием, должны быть специфицированы и включены в спецификацию стерилизационного процесса. Некоторые из примесей, подлежащих рассмотрению, и их максимально допустимые уровни приведены в приложении А.
6.1.2 Процессы с насыщенным паром
Пар может генерироваться внутри стерилизатора или впускаться в камеру из внешнего источника. Воздух может постепенно удаляться из камеры за счет гравитационного замещения, активного потока пара или за счет принудительного вакуумирования. Наличие насыщенного пара отмечается в точке измерения, например, в сливе камеры, когда измеренная температура совпадает с температурой насыщенного пара, рассчитанной по давлению (см. приложение С). И температура, и давление являются переменными процесса, а точка измерения температуры определяется как контрольная точка измерения.
Если колебания параметров процесса и/или объем неконденсируемых газов могут привести к неэффективному процессу, изготовитель стерилизатора или ответственное лицо (см. ИСО 17665-1:2006, приложение А, пункт А.4.2) обязан обеспечить пользователя адекватной информацией, которая должна включать в себя:
- верхний и нижний пределы для каждого параметра процесса, а также способ удаления воздуха из камеры;
- источники неконденсируемых газов;
- методы испытаний, частоту испытаний и критерии приемлемости для оценки процесса стерилизации.
Удаление воздуха из камеры стерилизатора за счет активного потока или гравитационного замещения имеет предсказуемый результат только при обработке простых твердых медицинских изделий. Удаление воздуха непредсказуемо для таких медицинских изделий как инструменты с каналами, большие твердые изделия с крупной массой, а также инструменты и текстильные изделия в своей первичной упаковке. Для обработки таких медицинских изделий должен использовать рабочий цикл с принудительным или динамическим способом удаления воздуха. Примером может служить цикл, использующий несколько вакуумных и/или паровых пульсов для последовательного уменьшения количества воздуха в камере и в медицинском изделии (изделиях). Во время каждой пульсации пар входит внутрь медицинского изделия и выходит из него; конденсирующийся пар вновь испаряется, и за счет этого обеспечивает динамическое "вымывание" остатков воздуха из упаковок, щелей и каналов. Количество пульсов, верхнее и нижнее значение давления в каждом пульсе, скорость изменения давления и температуры и интервал между каждым таким изменением являются переменными процесса и играют важную роль для эффективного удаления воздуха. При оценке пригодности процесса стерилизации к семейству продуктов комбинации этих изменений температуры и давления, скорости их изменения и продолжительность каждого такого изменения должны быть приняты во внимание.
Как только измеренная температура превысит теоретическую температуру, рассчитанную согласно описанию, приведенному в приложении С, уже может появиться перегретый пар. Наличие перегретого пара может быть пагубным для медицинского изделия и/или для его упаковки, и может также привести к неудачному процессу стерилизации.
Эффективное удаление воздуха из каналов инструментов, пористых загрузок и других изделий сложной конструкции, имеющих труднодоступные полости, достаточно затруднительно. На физические условия, требующиеся для эффективного удаления воздуха, влияют длина, ширина и форма каналов, толщина стенки изделия, материал продукта, масса, плотность, система упаковки и наличие других изделий в той же упаковке. Процесс стерилизации, успешно удаляющий воздух из камеры стерилизатора до минимального уровня, может оказаться неспособным удалить в достаточной степени воздух из канала изделия, чтобы позволить проникнуть туда пару. Закон Дальтона гласит, что суммарное давление в замкнутом пространстве равно сумме парциальных давлений различных газов, присутствующих в этом пространстве. Теоретически температура в камере стерилизатора, содержащей смесь пара и остаточного воздуха, должна быть ниже, чем расчетная температура, полученная от соответствующего значения давления в соответствии с таблицей значений температур пара для измеренных давлений в камере (см. приложение С).
Однако имеются факты, показывающие, что объем остаточного воздуха, достаточный для того, чтобы цикл стерилизации оказался неудачным, может понизить температуру пара всего лишь на такую малую величину, как 0,01°С. Вследствие этого разница между температурой, измеренной в контрольной измерительной точке, и расчетной температурой, полученной с помощью таблицы значений температур пара для измеренных давлений в камере (см. приложение С), может оказаться недостаточной для обнаружения небольших объемов воздуха, способных концентрироваться в каналах или труднодоступных полостях изделий, предотвращая проникание в них пара. При таких обстоятельствах адекватное удаление воздуха и проникание пара в загрузку может быть спрогнозировано по данным, полученным из испытания на проникание пара и/или по данным, полученным от устройства для мониторинга процесса.
Испытание на проникание пара предназначено для специфических семейств продуктов и используется для проверки того, что объем неконденсируемых газов, оставшихся в камере стерилизатора к началу периода плато, не воспрепятствует контакту насыщенного пара со всеми поверхностями медицинского изделия на протяжении времени выдержки. На этот объем влияют эффективность системы удаления воздуха, натекание воздуха в камеру стерилизатора и неконденсируемые газы, переносимые паром. Натекание воздуха в камеру и объем неконденсируемых газов, содержащихся в паре, могут быть определены с помощью различных испытаний (см., например, приложение А и ЕН 285). Общее наличие неконденсируемых газов определяют испытанием на проникание пара.
Испытание на проникание пара может быть основано на измерении температуры, на биологических или химических индикаторах в зависимости от возможностей. Испытательная система должна обеспечивать нагрузки, представительные для того семейства (семейств) продукта, для которого она предназначена. Выпускается целый ряд испытательных устройств для контроля проникания пара и удаления воздуха. Требования к химическим индикаторам можно найти в ИСО 11140-3, ИСО 11140-4, ИСО 11140-5, ИСО 11140-6 и ЕН 285.
_______________
ИСО 11140-6 находится в стадии разработки и основывается на ЕН 867-5 [57].*
* Вероятно ошибка оригинала. Следует читать: [26]. - .
Руководство по выбору и использованию химических индикаторов приведено в ИСО 15882. Требования к биологическим индикаторам приведены в ИСО 11138-3. Руководство по выбору и использованию биологических индикаторов находится в ИСО 14161.
Контрольная загрузка может состоять из единичного типа медицинского изделия, из медицинских изделий, относящихся к разным семействам изделий, но заключенным в единую общую упаковку. Для каждого контрольного продукта или медицинского изделия трудность удаления воздуха или иные помехи процессу стерилизации должны быть меньше, чем для любого медицинского изделия или семейств продуктов, для которых рассчитан данный процесс (см. также приложения А и В).
Если предполагается использовать устройство контроля процесса (например, детектор воздуха или иное устройство мониторинга) для представления семейства (семейств) продукта, пригодность данного устройства при экспозиции в условиях процесса стерилизации должна быть подтверждена изготовителем устройства для испытания процесса, изготовителем стерилизатора или ответственным лицом (см. ИСО 17665-1:2006, приложение А, пункт А.4.2).
6.1.3 Процессы с упакованными продуктами
Продукт может быть нагрет в цикле с погружением его в воду, в цикле с разбрызгиванием воды, в цикле с паровоздушной смесью, в паровом цикле с гравитационным замещением воздуха или в цикле с принудительным удалением воздуха. Паровоздушные смеси часто используются для предотвращения искажения геометрической формы или нарушения структуры стерилизуемого контейнера, возникающими из-за внутреннего давления, генерируемого за счет нагрева как растворов на водной основе, так и за счет наличия воздуха в любом герметично укупоренном контейнере. Энергия, необходимая для нагрева стерилизационной загрузки до определенной температуры стерилизации, зависит от семейства продукта, размера стерилизационной загрузки и ее начальной температуры. Перенос тепла будет зависеть от нагревающей среды, ее контакта с контейнером продукта, от материала контейнера и системы поддержки контейнера, а также от разницы температур в месте переноса тепла. Тип семейства продукта и конфигурация загрузки будут оказывать наибольшее влияние на разницы температур в месте переноса тепла. Эти разницы можно минимизировать путем уменьшения потока и улучшения распределения нагревающей среды за счет принудительной циркуляции. Массовый поток и гомогенность переносящей тепло среды в объеме камеры могут быть проверены с помощью таких переменных процесса, как скорость вращения вентилятора, давление и поток циркуляции. Температура переносящей тепло среды на ее выходе должна быть определена как переменная процесса. Если используется пар, температура окружающей среды также должна считаться переменной процесса. Может понадобиться уделить внимание апирогенности переносящей тепло среды и отсутствию в ней химических примесей, способных вызвать образование пятен на упаковке.
В дополнение к сказанному, может потребоваться, чтобы переносящая тепло среда была стерильной во время охлаждения и в течение периода рабочего цикла, в течение которого обеспечена летальность.
Распределение температуры внутри контейнера с продуктом будет зависеть от формы контейнера, вязкости продукта, условий передачи тепла через стенку контейнера и через продукт, а также от конвекции внутри продукта. Крупные контейнеры требуют большего времени для разогрева и охлаждения, что может вести к ограничению размеров контейнеров, используемых для продуктов, чувствительных к продолжительной экспозиции.
В ходе выполнения процесса стерилизации должны быть идентифицированы точки размещения контейнеров с продуктом, в которых измерены наивысшая и низшая температуры в загрузке во время фазы нагрева, а также наивысшая и низшая температуры в загрузке во время фазы охлаждения. Температуры, измеренные в этих точках, должны быть рассмотрены как переменные процесса; однако если любая из этих точек оказывается невоспроизводимой, для обеспечения специфицированной летальности с одновременной сохранностью продукта может понадобиться статистический подход.
6.2 Оборудование
Примечание - Особые соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.3.2.
6.2.1 Опубликованы региональные и национальные стандарты на стерилизационное оборудование (например, ЕН 285), рекомендующие материалы, которые могут быть использованы при конструировании и изготовлении стерилизаторов. Материалы, используемые изготовителем в конструкции стерилизатора, могут базироваться на процессе стерилизации, осуществляемом стерилизатором, и на семействе (семействах) продукта, которые предполагается в нем стерилизовать. Выбранные материалы должны минимизировать коррозию и образование любых контаминантов, которые могут выделяться в ходе рутинной эксплуатации. Пар, иная переносящая тепло среда, жидкости или воздух, используемые для опрессовки камеры стерилизатора, могут переносить вместе с собой токсичные или коррозийные агенты. Такие агенты должны быть идентифицированы, и для них должны быть оговорены максимально допустимые уровни (см. приложение А). Защита материалов за счет пленкообразующих аминов (таких как гидразин) не должна применяться в качестве альтернативы правильному подбору материалов и контролю коррозийных примесей.
Предпочтительно, чтобы записи о стерилизации были независимыми от автоматического контроллера и показывающих приборов стерилизатора. Система, комбинирующая в себе запись данных, управление и индикацию, может привести к неэффективному процессу стерилизации, который будет интерпретирован как эффективный. Независимые устройства регистрации данных характеризуются наличием собственных отдельных средств измерения, обработки данных и распечатки их значений. При этом не исключается обмен информационными данными в иных целях между регистратором и контроллером.
Воздушный детектор может быть встроен в стерилизатор, использующий ваккумные и паровые пульсы для откачки воздуха в фазе удаления воздуха процесса стерилизации насыщенным паром. Он используется для определения того, могут ли неконденсируемые газы, остающиеся в камере стерилизатора к концу периода плато, накапливаться в частях загрузки (например, в каналах) и приводить тем самым к неудачному завершению процесса стерилизации в этих частях. Уставка воздушного детектора основывается на определенных параметрах процесса и данных о семействе (семействах) продукта, для обработки которых разработан данный процесс. Неконденсируемые газы, обнаруженные детектором воздуха, могут в числе прочего содержать и газы, выделяющиеся при нагреве продукта или его упаковки. Испытания детектора воздуха изложены в приложении А и в ЕН 285.
6.2.2 Спецификации оборудования должны включать в себя информацию, достаточную для выполнения определения процесса для нового продукта или новой конфигурации загрузки (см. раздел 8).
6.2.3 Процесс стерилизации, выполняемый в соответствии с его спецификацией, зависит от качества обеспечивающих его питающих сред. В моменты пикового потребления давление каждой питающей среды - газа, жидкости или пара, замеренное в точке ее подключения к стерилизатору, не должно падать ниже минимума, специфицированного изготовителем стерилизатора.
Например, эффективность работы вакуум-насосов типа "водяное кольцо" и теплообменников падает вместе со снижением давления воды и с повышением ее температуры. Может появляться микробная контаминация, если впускаемый в камеру стерилизатора воздух содержит частицы размером более 2 микрометров. Если подача питающих сред обеспечивается третьими лицами, они обязаны следовать рекомендациям изготовителя стерилизатора, и соответствие этим рекомендациям должно быть подтверждено.
Локальные нормативные требования, относящиеся к охране окружающей среды, могут накладывать ограничения на высокую температуру жидкостей, истекающих из стерилизатора в общую канализационную систему, на уровни утечки материалов, используемых для производства стерилизующего агента, на частицы, выделяемые продуктом и/или упаковкой в ходе стерилизации, и на объем воды, используемой в ходе процесса.
Безопасность должна быть частью конструкции оборудования и работы с ним. Следует соблюдать требования МЭК 61010-2-040 и требования национальных нормативных документов.
6.2.4 Такие системы как контейнеры, полки, стеллажи и носители, предназначенные для поддержки и/или для содержания внутри себя медицинского изделия (изделий), не должны препятствовать единообразному распространению пара, циркуляции переносящей тепло жидкости, удалению остаточного воздуха, сливу конденсата или воды. Система должна также препятствовать повреждению медицинского изделия и/или его упаковки и сохранять целостность стерилизационной загрузки.
6.2.5 Нет дополнительной информации.
6.2.6 Программное обеспечение должно быть структурировано. Указания даны в издании "Правила надлежащего производства автоматики. Руководство по валидации автоматизированных систем в фармацевтической промышленности" (GAMP 4).
7 Определение продукта
Примечание - Особые соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, подраздел D.4.
7.1 В ходе разработки продукта следует уделять внимание процедурам его разборки (если это применимо), очистки, дезинфекции, осмотра и стерилизации.
Руководства и методы очистки и дезинфекции медицинских изделий перед стерилизацией рассмотрены в серии стандартов ИСО 15883. Информация об обработке медицинского изделия, которая должна предоставляться его изготовителем, дана в ИСО 17664.
7.2 Основной функцией упаковки является сохранение медицинского изделия стерильным, пока упаковка не будет вскрыта для использования изделия. Упаковка должна противостоять воздействиям, которые могут возникать в ходе процесса стерилизации, оставаться закрытой, и не должна оказывать негативного воздействия на качество медицинского изделия (например, за счет выделения ею частиц).
Упаковка медицинского изделия, стерилизуемого насыщенным паром, должна удовлетворять требованиям ИСО 11607.
Материал и конструкция непроницаемых упаковок (например, флаконов, ампул, эластичных пакетов) должны позволять передачу тепла к продукту и, если контейнер укупорен, укупорка должна оставаться закрытой и обеспечивать герметичность.
Вторичная упаковка должна защищать продукт во время обычной работы с ним и его распространения. Если вторичная упаковка должна подвергаться процессу стерилизации, она должна сохранять свою способность защищать продукт и не должна ухудшать свои качества из-за воздействия на нее процесса стерилизации.
Если в конце процесса стерилизации нужны контролируемые условия для уравнивания давления в медицинском изделии и его упаковке до атмосферного давления, то должен быть определен способ, с помощью которого эти условия достигаются (например, в камере или в комнате с регулируемыми условиями окружающей среды).
7.3 Нет дополнительной информации.
7.4 Подлежащее стерилизации медицинское изделие может быть охарактеризовано его формой, массой, материалами конструкции, наличием движущихся частей и упаковкой. Продукт в контейнере будет характеризоваться его составом, объемом и вязкостью. Его контейнер будет характеризоваться размером, материалом и укупоркой.
Следует выполнять исследование с целью отнесения продукта к определенному семейству продуктов. Объем такого исследования может быть сокращен за счет первого пересмотра параметров процесса, уже установленных для существующего процесса стерилизации, за счет применения валидированного процесса очистки (если это применимо), и за счет сравнения нового продукта с теми продуктами, которые уже соотнесены с процессом стерилизации [приписаны к процессу стерилизации].
7.5 Нет дополнительной информации.
7.6 Экспонирование медицинского изделия воздействию стерилизующего агента не должно позволять выход расчетных параметров любых используемых в конструкции изделия материалов за установленные для них максимальные или минимальные допустимые пределы.
По мере роста температуры материалы разупрочняются и становятся более чувствительными к физическим напряжениям или механическим воздействиям.
Дифференциальное расширение материалов с низкой теплопроводностью или расширение и сжатие различных материалов, находящихся в контакте между собой, могут стать причиной возникновения напряжений в материалах и швах изделий.
7.7 Нет дополнительной информации.
7.8 Теплочувствительность жидкого продукта может диктовать максимальный объем заполнения, материал и размер контейнера, которые можно использовать. Стабильность и стерильность жидкости должна быть оценена по исследованиям температурных карт, выполненных на предлагаемых контейнерах, когда жидкость подвергается воздействию по меньшей мере верхних пределов выбранного [предлагаемого] профиля процесса стерилизации.
Медицинские изделия, подвергающиеся повторной обработке, могут страдать от аккумулирующихся изменений, выражающихся в растрескивании поверхностей от дифференциального расширения толстых материалов, появлении хрупкости или расслаивания. Трещины, изломы, разрывы и каналы могут удерживать в себе органические, химические и биологические примеси [контаминанты], которые могут вызывать нежелательные [материальные] реакции или непредсказуемо выделяться наружу во время использования изделия. Многие материалы, подвергаясь многократному воздействию стерилизации влажным теплом, могут сохранять длительный срок безопасной службы (например, нержавеющая сталь). Другие материалы, однако, могут иметь ограниченный срок функционирования и требуют дополнительных исследований. Ссылки следует искать в ИСО 10993-1, ИСО 10993-17, ИСО 17664 и ИСО 14971.
7.9 Оценка должна установить, что после обработки медицинское изделие будет действовать по своему назначению и будет безопасно в использовании. Оценка должна учитывать механические, химические, электрические, токсикологические, физические, биологические и морфологические свойства изделия. Специальные добавки, процессные примеси [контаминанты], выщелачиваемые вещества, остаточные субстанции процесса и продукты деградации должны быть оценены по степени их воздействия на безопасность изделия и его упаковки. Может проявляться коррозия некоторых материалов, если пар образуется из воды с низким рН, или из воды, содержащей такие примеси, как хлориды и силикаты. Например, резина может оксидироваться в присутствии остаточного воздуха при пониженных температурах пара. Обезвоженные целлюлозные материалы могут набирать воду в ходе процесса паровой стерилизации, вызывая тем самым экзотермический перегрев материала и пространства вокруг него.
7.10 Нет дополнительной информации.
8 Определение процесса
Примечание - соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, подраздел D.5.
8.1 Целью этого действия является обеспечение требуемого уровня обеспечения стерильности в каждой части загрузки стерилизатора за счет гарантированного контакта контаминирующих микроорганизмов с влагой при заданной температуре в течение заданного времени.
Эффективность и воспроизводимость стерилизационного процесса могут определяться условиями, которые можно контролировать и подтверждать с помощью физических измерений. Если одно из условий изменяется, и это может повлиять на уровень обеспечения стерильности, такое условие должно быть определено как переменная процесса, а значение, при котором возникает изменение, должно быть определено как параметр процесса.
Переменные процесса должны быть определены, а параметры процесса, в том числе и их допуски, должны быть специфицированы. Параметры процесса должны характеризовать условия, подтверждающие прогноз того, что стерилизующий агент обеспечит требуемый уровень обеспечения стерильности во всех частях продукта, не позволяя при этом ни одной его части выйти за расчетные допустимые пределы.
При обработке некоторых медицинских изделий измерение физических условий (например, температуры) внутри систем стерильного барьера не представляется возможным. Для таких медицинских изделий воспроизводимое достижение заданного уровня обеспечения стерильности должно быть верифицировано в контрольных измерительных точках, например, в сливе для измерения температуры стерилизации. В случае использования процесса с насыщенным паром доказательства воспроизводимости процесса могут быть получены из:
a) значений температуры и давлений, по меньшей мере в точках перегибов графика давления;
b) количества паровых пульсов;
c) скоростей изменения давления и/или температуры;
d) длительности экспозиции;
e) величины натекания воздуха в камеру;
f) данных о качестве пара.
Процесс стерилизации может быть разработан в производственном стерилизаторе или в специальном исследовательском стерилизаторе. Параметры процесса для определенного процесса стерилизации должны быть установлены на их наихудшие (наименьшие допустимые) значения, но в то же время еще приемлемые для эффективной стерилизации, например, с использованием нижнего предела допуска на длительность экспозиции или с использованием наименьшей допустимой скорости рециркуляции для процесса с погружением изделия в воду.
8.2 Иногда для обработки продуктов, производимых медицинской и фармацевтической промышленностью, выбирается процесс, основанный на рекомендациях по температуре стерилизации и времени выдержки, приведенных в региональной или национальной фармакопее.
8.3 Нет дополнительной информации.
8.4 Нет дополнительной информации.
8.5 Биологический индикатор представляет собой микробиологическую нагрузку с известной устойчивостью (резистентностью), используемую для подтверждения летальности стерилизационного процесса в определенных местах камеры или в продукте (см. ИСО 11138-1) в точках размещения индикаторов.
Разработка и определение микробиологических процессов рассматриваются в приложении В и ИСО 17665-1:2006, приложения В, С и D. При использовании биологических индикаторов следует уделять внимание микроорганизмам, удерживаемым внутри продукта, примесям (контаминантам) внутри или на поверхностях продукта, нежелательным реакциям с материалами конструкции, а также учитывать трудность размещения биологических индикаторов в полостях и каналах изделий.
Когда бы ни использовались биологические индикаторы для подтверждения летальности в заданных точках их размещения, всегда должно быть использовано измерение физических параметров в ходе процесса стерилизации с целью подтверждения того, что выполнялся именно заданный стерилизационный процесс.
8.6 Нет дополнительной информации.
8.7 Нет дополнительной информации.
8.8 В качестве одного из элементов в определении стерилизационного процесса может быть использован химический индикатор (см. ИСО 11140-1).
Он используется для демонстрации достижения заданных величин переменных процесса в месте своего размещения.
Химические индикаторы показывают экспозицию за счет воздействия на них изменяющихся физических и/или химических факторов, и рассчитываются на реакцию на изменение одного или нескольких переменных стерилизационного процесса, таких, например, как длительность экспозиции, температура или влажность. Изготовитель химического индикатора должен определить условия экспозиции, которые могут заставить химический индикатор изменить свое начальное состояние на конечное. Достижение химическим индикатором конечного состояния не должно рассматриваться как индикация достижения приемлемого уровня обеспечения стерильности, но может быть одним из многих факторов, учитываемых при оценке приемлемости процесса стерилизации. Отсутствие перехода химического индикатора в конечное состояние должно рассматриваться как доказательство неудачного выполнения процесса стерилизации, и такой случай подлежит расследованию. Руководство по использованию химических индикаторов приведено в ИСО 15882.
8.9 При разработке процесса могут быть использованы данные, полученные от контрольного устройства, такого, например, как устройство контроля процесса и/или контрольного устройства, предназначенного для имитации свойств продукта или семейства продуктов. Для процессов с использованием насыщенного пара рассмотрения требуют следующие факторы:
- материалы конструкции;
- масса;
- длина и диаметр полых изделий и трубок;
- способность к впитыванию (поглощению) влаги;
- теплопроводность;
- границы безопасности, связанные с нагрузкой;
- средства, с помощью которых могут оцениваться удаление воздуха и проникание пара.
Эти факторы обычно могут быть оценены путем измерения температуры в сочетании с использованием химических и/или биологических индикаторов.
Для продуктов в контейнерах контрольное устройство должно имитировать температурный профиль в наихудшем месте размещения внутри продукта.
8.10 Если продукт был приписан к одному из семейств продуктов, для которого процесс стерилизации уже определен, и этот процесс основан на установленном соотношении времени/температуры, то дополнительная оценка с помощью биологических индикаторов обычно не обязательна.
8.11 Процесс стерилизации, базирующийся на определенной биологической нагрузке, представленной биологическими индикаторами, используется при разработке стерилизаторов в пищевой, фармацевтической промышленности, в медицинской промышленности и в учреждениях здравоохранения. Этот метод известен как метод "полной гибели" (см. ИСО 17665-1, приложение D).
8.12 Процесс стерилизации, базирующийся на биологической нагрузке в ее естественном состоянии или в сочетании с биологическими индикаторами, требует расширенных биологических исследований, сопровождаемых частым биологическим скринингом продукта и окружающей среды.
Этот метод обычно используют в фармацевтической и медицинской промышленности. Его выбирают, если во время определения продукта некоторые свойства продукта или оборудования продемонстрировали свою чувствительность к обработке влажным теплом. В таком случае используют минимальный процесс для достижения условий, при которых продукт может быть назван "стерильным" без ухудшения его качества или функций (см. ИСО 17665-1, приложения В и С). Испытания на наличие жизнеспособных микроорганизмов должны быть выполнены на стерилизованных носителях с известной популяцией микроорганизмов, имеющих известную устойчивость (резистентность).
8.13 Нет дополнительной информации.
9 Валидация
9.1 Общая информация
9.1.1 Новый стерилизатор должен быть поставлен и смонтирован в соответствии с его спецификациями и монтажными чертежами.
Должно быть возможно перемещение элементов валидации между аттестацией монтажа (IQ), аттестацией функционирования (OQ) и аттестацией эксплуатируемого оборудования (PQ), если при планировании специфической валидации это окажется более практичным действием.
Документированный валидационный план должен быть согласован и утвержден ответственными сторонами прежде, чем начнутся валидационные исследования. Валидационные документы должны проходить через процедуры регистрации их истории и контроля изменений (см. ИСО 17665-1, пункт 9.1.3).
9.1.2 Нет дополнительной информации.
9.1.3 См. пункт 9.1.1, параграф 3.
9.1.4 Цепи измерения температуры должны быть верифицированы с помощью контрольно-измерительных калибровочных средств измерения и действующих стандартов. Одним из примеров может служить масляная ванна с известной стабильной температурой, контролируемая образцовым измерителем температуры, соответствующим стандарту. При одновременном погружении в ванну нескольких датчиков температуры может быть определена разность их показаний (т.е. погрешность датчиков при измерении заданной температуры).
Если разности между измеренными температурами используются для оценки результатов процесса стерилизации, то должна быть известна погрешность каждого измерения при том значении температуры, при котором должно делаться сравнение.
9.1.5 Калибровка приборов, встроенных в стерилизатор, и калибровка измерительных цепей, используемых для контроля и управления, может быть подвергнута частой верификации в критических частях рабочего цикла путем сравнения с измерениями, сделанными контрольными приборами во время выполнения рабочих испытаний
9.1.6 См. пункт 9.2.3, параграф 1.
9.1.7 Нет дополнительной информации.
9.1.8 См. приложение D, пункты D.6.2.3 и D.6.3.
9.2 Аттестация монтажа (IQ)
9.2.1 Оборудование
Аттестация монтажа необходима при вводе в эксплуатацию нового стерилизационного отделения, при замене имеющегося стерилизатора на новый, или при перемещении имеющегося стерилизатора на новое место. Некоторые элементы аттестации монтажа необходимо выполнить, если в имеющийся стерилизатор вносятся изменения, способные повлиять на эффективность процессов стерилизации, например, заменяются дверные прокладки, модифицируется система подачи пара, заменяется или ремонтируется вакуум-насос.
План выполнения аттестации монтажа, который может составлять часть мастер-плана валидации, должен включать в себя процедуры, обеспечивающие получение документированных доказательств того, что стерилизатор и сопровождающая документация соответствуют спецификациям.
Изготовитель стерилизатора обязан предоставить руководство по проведению испытаний и текущего контроля каждой системы обнаружения неполадок как часть сопроводительной документации стерилизатора, например, должны быть приведены методы имитации неполадок в системе подачи питающих сред или создания объема остаточного воздуха в камере.
9.2.2 Монтаж
План аттестации должен включать в себя процедуры, обеспечивающие документированное свидетельство того, что все присоединенные к стерилизатору питающие среды соответствуют спецификациям, и что рабочий цикл соответствует циклу, специфицированному изготовителем.
9.2.3 Функционирование
После монтажа должны быть установлены и подтверждены функции систем безопасности согласно требованиям МЭК 61010-2-40.
План аттестации должен включать в себя процедуры, обеспечивающие получение документированных доказательств того, что не обнаружено никаких признаков неисправностей или утечек, и что в моменты пикового потребления давления всех питающих сред превышают или равны минимально допустимому значению, специфицированному изготовителем стерилизатора.
Следует верифицировать функционирование систем обнаружения неисправностей, встроенных в стерилизатор изготовителем.
На этой стадии или во время выполнения аттестации функционирования можно выполнить верификацию калибровки измерительных систем, встроенных в стерилизатор, а также проверку каждой системы, используемой для регистрации или идентификации неполадок, не позволяющих достигнуть заданных значений критических параметров процесса.
9.3 Аттестация функционирования (OQ)
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.6.1.
9.3.1 План аттестации функционирования, который может составлять часть мастер-плана валидации, должен включать в себя процедуры, обеспечивающие получение документированных доказательств того, что:
а) системы безопасности и обнаружения неполадок, встроенные в стерилизатор, функционируют в соответствии со спецификацией;
b) установленное оборудование работает в заранее заданных пределах;
c) качество каждой питающей среды соответствует спецификации;
d) рабочий цикл работает в соответствии со спецификацией;
e) в ходе выполнения рабочего цикла не обнаружено свидетельств воздействия стерилизатора на окружающее оборудование и наоборот;
f) звуковое давление на месте установки стерилизатора не превышает региональные или национальные нормативные требования;
g) при работе стерилизатора с пустой камерой температура и давление, записываемые и показываемые встроенными приборами на протяжении всего цикла стерилизации, находятся в заданных пределах для стерилизационного процесса;
h) отсутствуют очевидные утечки пара, сжатого воздуха, воды или сбросов при любой температуре или давлении в пределах рабочего диапазона стерилизационного цикла.
Максимальное и минимальное значения любой процессной переменной не должны превышать допустимых значений, указанных изготовителем медицинского изделия.
Если изготовителем стерилизатора рекомендованы испытания эксплуатируемого оборудования, они должны быть выполнены на стадии аттестации эксплуатируемого оборудования, при этом должно быть верифицировано соответствие критериям приемлемости, специфицированным изготовителем стерилизатора.
Если изготовитель декларирует соответствие стандарту на оборудование, испытания, проводимые в ходе аттестации функционирования, должны соответствовать испытаниям, специфицированным в стандарте на оборудование.
Если должен использоваться существующий процесс стерилизации, его текущий статус должен быть верифицирован путем демонстрации соответствия результата текущих испытаний результатам предыдущих испытаний, проведенных в ходе аттестации монтажа и аттестации функционирования.
Для процессов с использованием насыщенного пара:
- качество пара и натекание воздуха в камеру стерилизатора могут влиять на эффективность процесса стерилизации. Должно быть продемонстрировано соответствие требованиям ИСО 17665-1, разделы 7 и 8 и/или рекомендациям изготовителя медицинского изделия;
- если требуется выполнение испытания на проникание пара (см. ИСО 17665-1, раздел 6), должно быть продемонстрировано соответствие требованиям к качеству работы и требованиям к проведению испытаний;
- если испытание на проникание пара должно выполняться рутинно для проверки удаления воздуха и проникания пара, то валидность испытания должна быть подтверждена, например, соответствием требованиям стандартов, описывающих эти испытания, таких как ИСО 11140-3, ИСО 11140-4 или ИСО 11140-5;
- если для представления специфического продукта должно быть использовано устройство контроля процесса, то процесс стерилизации должен быть проведен с этим устройством. Оно может быть использовано отдельно или быть включено в другие испытания. Следует выполнять инструкции, предоставленные изготовителем устройства контроля процесса;
- если для текущего контроля должен быть использован воздушный детектор, он должен быть отрегулирован [настроен] в ходе испытаний аттестации функционирования с использованием контрольной загрузки. Воздушный детектор должен подавать сигнализацию о неполадке, если в ходе удаления воздуха в контрольной загрузке не были достигнуты заданные значения переменных процесса. Контрольная загрузка должна быть представительной для "худшего случая" медицинского изделия и конфигурации загрузки.
- Приложение А определяет испытания, которые должны быть выполнены со стерилизационным процессом в ходе аттестации функционирования с использованием параметрического подхода. Приложение В определяет испытания, которые должны быть выполнены со стерилизационным процессом в ходе аттестации функционирования с использованием биологического подхода.
- Если уровень остаточной влажности продукта может повлиять на его качество при использовании по назначению (например, облегчит его микробную ре-контаминацию), то должно быть выполнено испытание на влажность загрузки.
Для продуктов в контейнерах:
- профили нагрева, экспозиции и охлаждения должны быть проверены при пустой камере;
- должны быть определены "холодное пятно" и "горячее пятно";
- должно быть верифицировано соответствие требованиям таких переменных процесса, как давление насоса, циркуляция и температура.
9.3.2 Нет дополнительной информации.
9.4 Аттестация эксплуатируемого оборудования (PQ)
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.6.2.
9.4.1 Целью аттестации эксплуатируемого оборудования является демонстрация способности стерилизационного процесса достигать заданного уровня обеспечения стерильности конкретной загрузки на повторяющейся основе.
Следует иметь план аттестации эксплуатируемого оборудования, который может быть частью валидационного мастер-плана.
9.4.2 Нет дополнительной информации.
9.4.3 Должны быть включены процедуры, обеспечивающие документированное свидетельство того, что стерилизационный процесс будет стерилизовать продукт (продукты), приписанный к семейству продуктов, для стерилизации которого данный процесс разработан.
Если изготовителем стерилизатора рекомендован предварительный разогрев камеры перед использованием, это должно быть указано и исполнено перед выполнением испытаний аттестации эксплуатируемого оборудования.
Загрузка стерилизатора и ее конфигурация должны быть такими, какие будут использоваться в рутинной работе. Если предусматривается повторная обработка, при испытаниях должны быть использованы наихудшие возможные конфигурация загрузки и комбинация продуктов из семейств, предназначенных для обработки данным процессом. Упаковка также должна быть той, что будет использоваться в рутинной работе.
Стерилизованные продукты, прошедшие через процесс заключительной стерилизации, должны быть подвергнуты проверке на стерильность. Проверки на стерильность имеют невысокую статистическую релевантность и не могут быть приняты в качестве единственного доказательства валидности процесса стерилизации.
9.4.4 Для процессов с использованием насыщенного пара:
- Качество пара и натекание воздуха в камеру стерилизатора могут пагубно воздействовать на заданные переменные процесса, и они должны быть обнаружены перед выполнением испытаний аттестации эксплуатируемого оборудования. Если должно использоваться испытание на проникание пара, например, тест Бови-Дика, результаты его выполнения должны быть известны заранее.
- Действенность удаления воздуха и проникания пара, косвенным путем определенные из результатов испытаний, установленных для аттестации монтажа и аттестации функционирования, должны быть проверены и верифицированы для "худшего случая" в отношении загрузки, ее конфигурации и медицинских изделий. Данные, на основании которых выносится суждение, должны быть получены из измерений температуры, поддержанных химическими и/или биологическими индикаторами, размещенными в трудно поддающихся стерилизации местах. Если должны быть использованы контрольная загрузка и/или устройство контроля процесса в качестве альтернативы "худшему случаю" загрузки, следует предварительно установить их валидность в качестве наибольшей нагрузки. Типы медицинских изделий и конфигураций загрузки, представляемые этими альтернативными средствами, могут быть указаны изготовителем стерилизатора. Устройство контроля процесса должно быть упаковано в тот же тип упаковки с использованием тех же процедур, что и продукт при рутинной работе. Одно и то же устройство, но с различными системами упаковки (например, пакеты или контейнеры), может представлять различные семейства продуктов.
- На протяжении этапов удаления воздуха и выравнивания стерилизационного цикла разность между температурами, измеренными в контрольной точке измерения и в точках медицинского изделия или контрольной загрузки, может быть использована для определения присутствия насыщенного пара в точках измерения. Период плато представляет собой комбинацию времени выравнивания и времени выдержки. В большинстве случаев время выдержки является частью рабочего цикла, обеспечивающей летальность.
- Для загрузок, определенных в приложении А, время выравнивания является мерой остаточных неконденсируемых газов, присутствующих в начале периода плато. Увеличение объема этих газов будет уменьшать время выдержки и снижать летальность. Скорость роста давления от точки вакуума до точки начала периода плато может влиять на определение наличия насыщенного пара. Низкая скорость роста давления может приводить к разогреву остаточного воздуха, что уменьшит разницу температур и приведет к неправильной интерпретации записанных данных в отношении проникания пара.
- Проникание тепла в каждый тип стерилизуемой загрузки должно быть определено либо по температуре, измеренной в определенном количестве упаковок с медицинскими изделиями, либо в контрольной загрузке. По меньшей мере один датчик температуры должен быть расположен в непосредственной близости от датчиков температуры, подключенных к записывающему устройству, показывающему прибору и контроллеру. Если датчик или индикатор не могут быть установлены на медицинском изделии в том месте, которое считается наиболее трудным для стерилизации, медицинское изделие может быть заменено на изделие другого типа или на устройство для испытания эффективности процесса при условии, что подтвержденная нагрузка на процесс от этой альтернативной замены должна быть равной или большей, чем от медицинского изделия, которое замена должна представлять. Количество и точки размещения используемых датчиков будут зависеть от типа стерилизационной загрузки и размеров камеры стерилизатора. Датчики, размещаемые в загрузке, должны быть размещены на тех частях (или внутри частей) медицинского изделия, из которых труднее всего удаляется воздух. Следует с осторожностью интерпретировать термометрические данные, полученные изнутри полых или пористых медицинских изделий, способных удерживать внутри себя воздух. Измерения температуры сами по себе не могут показать различия между температурой горячего воздуха или температурой насыщенного пара. Наличие насыщенного пара может быть оценено по скорости подъема давления и росту температуры, а при необходимости - и по экспонированным химическим или биологическим индикаторам.
- Воспроизводимость в границах допустимых пределов должна быть проверена с использованием не менее трех повторных одинаковых циклов (см. ИСО 17665-1).
- Для процессов с продуктами в контейнерах испытательная загрузка и ее размещение в камере стерилизатора должны быть проверены по меньшей мере в местах, расположенных вблизи от контейнеров, определенных в ходе аттестации функционирования как получающие наименьшую и наибольшую экспозиции.
Соответствие критическим переменным, определенным в разделе 8, должно быть верифицировано. Если для нового семейства продуктов и/или новой конфигурации загрузки должен быть использован уже имеющийся процесс стерилизации, пределы экспозиции, определенные в разделе 7, должны быть пересмотрены, а достижение микробиологической эффективности, определенное в разделе 8, должно быть верифицировано. Если параметры процесса изменяются в ходе последующей разработки, следует верифицировать микробиологическую эффективность и пределы экспозиции для существующего семейства (семейств) продуктов.
9.4.5 Нет дополнительной информации.
9.4.6 Нет дополнительной информации.
9.4.7 Нет дополнительной информации.
9.5 Анализ и утверждение валидации
9.5.1 Данные, собранные в ходе валидации, должны быть пересмотрены и утверждены назначенным для этого лицом, организационно независимым от лиц, выполнявших испытания, готовивших отчет о валидации, и от лиц, ответственных за производство.
9.5.2 Данные, используемые для подтверждения процесса стерилизации, должны включать в себя (по мере применимости):
a) спецификацию стерилизатора и любые последующие ее изменения;
b) место размещения и уникальный идентификатор стерилизатора, например, его заводской номер вместе с названием, адресом изготовителя, типом стерилизатора и ссылкой на модель;
c) документацию, подтверждающую соответствие спецификациям по безопасности;
d) документы на сосуд, работающий под давлением;
e) руководство по обслуживанию и график планового обслуживания стерилизатора;
f) инструкцию по монтажу;
g) инструкцию оператора;
h) копии деклараций (сертификатов) в соответствии с требованиями нормативных документов, относящихся к медицинским изделиям (если применимо);
i) валидационный мастер-план, валидационный протокол и отчет о валидации вместе с документированными данными;
j) стандартные рабочие процедуры обслуживания, проверок и испытаний;
k) подробные сведения о любых изменениях, внесенных в стерилизатор, в его приборы или в систему управления;
I) подробные сведения обо всех обнаруженных неисправностях и мерах по их устранению;
m) описания конфигурации загрузки для каждого типа загрузки стерилизатора/семейства продуктов;
n) для продуктов в контейнерах и, если применимо, упакованных продуктов (например, продуктов в таре) - результаты испытаний на проникание пара для каждого типа загрузки/семейства продуктов;
о) параметры, используемые для контроля цикла стерилизации, и копию спецификации процесса стерилизации;
р) идентификацию всего персонала, участвующего в валидации, вместе с данными о профессиональной квалификации (в части, касающейся их способности выполнять работы):
- программа переаттестации, периодических и рутинных проверок/испытаний;
- обучающие руководства для рутинного эксплуатационного персонала;
- подтверждение, что стерилизатор смонтирован и подключен к питающим средам в соответствии со спецификацией;
- подтверждение, что калибровка испытательного оборудования была верифицирована, и что каждая измерительная цепь была откалибрована и поверена, и (при необходимости) были проведены регулировки;
- подтверждение, что оборудование было проверено, и что оно выполняет определенный процесс стерилизации с приемлемой воспроизводимостью;
- параметры процесса (включая их допуски), используемые для оценки возможности выпуска продукта;
- значение установки воздушного детектора и/или интерпретация биологического индикатора, используемого в одиночку или в сочетании с устройством для испытания эффективности процесса;
- для оборудования, уже находящегося в эксплуатации - результаты обслуживания и подтверждение, что данные, полученные из рутинной эксплуатации, являются удовлетворительными.
10 Текущий мониторинг и контроль
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, подраздел D.7.
10.1 Должна иметься утвержденная программа рутинного контроля и управления. Данные, полученные из контроля и управления, должны документироваться, пересматриваться, утверждаться и сохраняться.
10.2 Лица, ответственные за стерилизацию, должны убедиться в том, что плановое обслуживание было успешно и полностью выполнено перед пуском стерилизатора в ежедневную эксплуатацию. Кроме того, должно иметься свидетельство, что в отчеты об аттестации функционирования/периодической реаттестации включены те стерилизационные загрузки/семейства продуктов, которые предстоит стерилизовать.
10.3 Смотрите приложение D, пункт D.7.2, в котором приведены соображения, относящиеся к учреждениям здравоохранения.
10.4 Профили температуры и давления в камере могут генерироваться электронным способом или путем оценки записей значений температуры и давления, полученных в ходе выполнения цикла стерилизации. Затем эти профили могут быть использованы для сравнения с профилями, полученными в ходе валидации.
Если для текущего контроля должен быть использован биологический и/или химический индикатор, он должен быть размещен в месте (местах), которые по результатам валидации признаны наихудшими для создания условий стерилизации. Как альтернатива, индикаторы могут быть помещены в места, которые в результате использования одного из методов, описанных в ИСО 17665-1, приложения С или D признаны нагрузкой на стерилизационный процесс, достаточной для обеспечения летальности в наименее доступной части продукта.
10.5 Измерения температуры стерилизации, периода плато и давления в камере стерилизатора являются достаточными при работе с неупакованными медицинскими изделиями, стерилизуемыми насыщенным паром. Если медицинское изделие упаковано и/или неконденсируемый газ может быть удержан в таких частях изделия, как каналы, трубки или расщелины, следует использовать ежедневное испытание на проникание пара. Для доказательства наличия насыщенного пара и обнаружения условий образования перегретого пара в объеме камеры в ходе процесса может оказаться необходимым сравнение измеренной и расчетной температур. Если медицинское изделие упаковано и/или неконденсируемый газ может быть удержан в таких частях изделия, как каналы, трубки или расщелины, значение , рассчитанное по температуре в камере, уже не будет представлять летальность процесса, а потому не может быть использовано для оценки результатов стерилизационного процесса для данного типа медицинского изделия.
В дополнение к измерению параметров процесса, для рабочего цикла должно быть доказано проникание пара в загрузку, например, с помощью встроенного в стерилизатор детектора воздуха и/или устройства контроля процесса. И детектор воздуха, и устройство контроля процесса должны быть верифицированы и признаны годными для продукта в стерилизационной загрузке.
10.6 Температура жидкости в контрольных контейнерах, находящихся в местах, признанных наиболее холодными и наиболее горячими частями стерилизационной загрузки по результатам ряда выполненных исследовательских циклов, может использоваться для прогнозирования наиболее низкой и наиболее высокой температуры загрузки, состоящей из жидкостей. Иногда для прогнозирования воспроизводимого температурного профиля наиболее холодного продукта могут использоваться температурные профили, сгенерированные для камеры стерилизатора и циркулирующей переносящей тепло среды. Если необходимо измерять температуру в контрольных контейнерах, расположенных внутри стерилизуемой загрузки, можно рассмотреть применение беспроводных систем измерения.
10.7 Нет дополнительной информации.
11 Выпуск продукции после стерилизации
11.1 Результаты плановых периодических испытаний должны быть отмечены в документации по выпуску продукции.
Выпуск продукта может быть основан на сравнении температурного профиля стерилизационной камеры с температурным профилем, измеренным либо в контрольном продукте (продуктах), либо в месте (местах), в котором температурный профиль внутри продукта может быть спрогнозирован. Для выпуска продукта могут быть также использованы данные о достижении специфицированных значений температуры стерилизации, периода плато и полосы температур стерилизации в месте, в котором время выдержки может быть спрогнозировано.
Для медицинских изделий, стерилизуемых насыщенным паром, выпуск, основывающийся исключительно на температуре стерилизации и времени выдержки, должен быть ограничен только не упакованными медицинскими изделиями.
Если химические и/или биологические индикаторы используются рутинно, они должны считаться частью критериев выпуска и должны быть дополнением к измерению физических параметров.
Целостность упаковок и контейнеров должна проверяться визуально после выгрузки продукта из стерилизатора. Поврежденные упаковки и контейнеры должны считаться не соответствующим требованиям продуктом. Аналогично этому требованию должна иметься еще действующая система, соответствующим образом отбраковывающая влажные упаковки с целью предотвращения попадания реконтаминированных продуктов в распределительную сеть. Сушка должна быть выполнена в среде с контролируемым количеством частиц и микробных контаминантов. Для этой цели могут подходить помещения класса 7 согласно определению ИСО 14644-1 [14] .
11.2 Идентификация стерилизованных и необработанных продуктов может быть осуществлена с помощью физических барьеров, проходных стерилизаторов и индикаторов стерилизации на упаковках.
12 Поддержание эффективности процесса
12.1 Демонстрация непрерывной эффективности
Во всех случаях, когда записи текущего контроля, периодических испытаний и аттестации функционирования указывают на неприемлемые отклонения от данных, полученных при валидации, причина этих отклонений должна быть идентифицирована, а стерилизатор подвергнут переаттестации.
Если стерилизатор используется редко, периоды бездействия могут приводить к изменениям в его работе или к изменениям питающих его сред. Это, в свою очередь, может приводить к выполнению процесса, не соответствующего специфицированному процессу. Если стерилизатор периодически простаивает, следует выполнить критический пересмотр данных с целью установления влияния простоев на эффективность процесса и определения мер, которые следует предпринять для уточнения рутинного мониторинга, испытаний или необходимости переаттестации, подтверждающих эффективность процесса. Например, следует рассмотреть возможные последствия еженедельного отключения стерилизатора на выходные дни или влияние на эффективность процесса энергосберегающих систем.
12.2 Повторная калибровка
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.9.1.
Интервал между повторными калибровками каждой измерительной цепи не должен превышать 12 месяцев и может быть уменьшен, если выполнялось внеочередное обслуживание или были получены доказательства неточности измерений.
12.3 Техническое обслуживание оборудования
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.9.2.
12.3.1 Стерилизатор должен быть периодически проверен с целью подтверждения того, что он по-прежнему продолжает соответствовать спецификации, и что не обнаружено свидетельств его неправильной работы. Должны также быть выполнены проверки и испытания для доказательства того, что оборудование по-прежнему безопасно в эксплуатации (см. МЭК 61010-2-040 [24]), и что все питающие среды соответствуют требованиям.
12.3.2 Должна быть разработана схема технического обслуживания на базе рекомендаций изготовителя стерилизатора, изготовителя (изготовителей) средств измерения, а также на базе данных, полученных из рутинных работ по обслуживанию, испытаний, выполненных на заводе-изготовителе и опыта, полученного в ходе эксплуатации. Для каждого стерилизатора должен быть разработан набор процедур, содержащий полные инструкции для выполнения каждой задачи, входящей в техническое обслуживание. Схема техобслуживания и частота исполнения каждой входящей в него задачи должны быть основаны на рекомендациях, представленных изготовителем стерилизатора, эксплуатационной нагрузке на стерилизатор и на соображениях безопасности.
12.3.3 После завершения каждого технического обслуживания должны выполняться проверки оборудования и его функций.
12.4 Переаттестация
Примечание - Соображения, относящиеся к учреждениям здравоохранения, приведены в приложении D, пункт D.9.3.
Переаттестация выполняется с целью подтверждения того, что изменения не ухудшили эффективность процесса стерилизации, и что данные, полученные в ходе валидации, по-прежнему валидны. Для защиты от недокументированных изменений охват и интервал между каждой частью переаттестации должны определяться по типу данных процесса, полученных из периодических испытаний, и из данных, верифицирующих рутинную воспроизводимость параметров процесса. Как правило, переаттестация выполняется ежегодно.
Охват работ по переаттестации будет зависеть от причин, вызвавших несоответствие в работе; если была выполнена замена компонента (см. ИСО 17665-1, пункт 12.5) или внесены изменения в систему управления, то может быть достаточно доказать повторяемость аттестованного стерилизационного цикла. Если же, в случае исполнения процесса с упакованными изделиями или с пористой загрузкой, неисправность заключалась в натекании воздуха в камеру, то может оказаться необходимым только повторить испытание камеры на утечку, а затем выполнить испытание на проникание пара.
Переаттестация эксплуатируемого оборудования может понадобиться также в случае смены продукта, его упаковки или конфигурации загрузки, или когда данные о стерилизуемой загрузке не укладываются в специфицированные пределы.
Если в ходе переаттестации используются биологические индикаторы, их характеристики должны быть идентичны тем индикаторам, которые использовались в ходе валидации. Любые изменения, способные вызывать сомнения относительно эффективности процесса стерилизации, должны инициировать процедуру пересмотра.
Для облегчения сравнения данных аттестации эксплуатируемого оборудования и переаттестации считается нормальным использовать один и тот же формат отчета.
12.5 Оценка изменений
Нет дополнительных указаний.
Приложение А
(справочное)
Оценка процесса стерилизации, базирующаяся в первую очередь на измерении физических параметров
А.1 Введение
А.1.1 Оценка процесса стерилизации этим методом проводится обычно на основании данных, полученных в результате выполнения серии последовательных испытаний. Каждое испытание должно разрабатываться с целью идентификации факта достижения одного или нескольких параметров, специфицированных для стерилизационного процесса.
Испытания и требования к рабочим характеристикам, детализированные в данном приложении, являются примерами и относятся к стерилизаторам, соответствующим требованиям ЕН 285 [25] и применимы при условии, что используемые испытательное оборудование и процедуры также отвечают требованиям ЕН 285 (см. также ИСО 17665-1, пункт 9.1.4). Требования к испытаниям и рабочим характеристикам для малых стерилизаторов даны в ЕН 13060 [38].
Стерилизаторы, соответствующие требованиям ЕН 285 или ЕН 13060, предназначены в первую очередь для использования в системе здравоохранения, однако они могут использоваться и в производстве медицинских изделий.
А.1.2 Для стерилизаторов, не соответствующих ЕН 285 или ЕН 13060, может быть невозможно достижение критериев приемлемости, приведенных в данном приложении. Для таких стерилизаторов документированные валидационные процедуры могут включать в себя испытания и процедуры как из данного приложения, так и из приложения В. Полученные в ходе испытаний данные могут быть затем использованы для верификации эффективности рассматриваемого стерилизационного процесса, предназначенного для обработки медицинского изделия (изделий). Этот подход может также оказаться подходящим при демонстрации соответствия требованиям законодательства о медицинских изделиях (если необходимо).
А.1.3 При выборе испытательных приборов для валидационных исследований и рутинных испытаний следует уделять внимание количеству и типам необходимых сигнальных входов. Должны записываться данные температуры и давления. Могут понадобиться и другие входные сигналы. Например, испытания малых загрузок (см. А.4) требует использования как минимум семи сигнальных входов для датчиков температуры и одного сигнального входа для датчика давления.
А.2 Испытания полой загрузки
А.2.1 Это испытание на проникание пара внутрь медицинского изделия (изделий), содержащего каналы. Испытание основано на контрольном изделии, представляющем полую загрузку, изложенным в ЕН 285, приложение А, подраздел А.1. Это испытание дополняет другие испытания, в которых должна использоваться стандартная контрольная упаковка (см. А.3).
А.2.2 Результат испытания с полой загрузкой оценивается по экспозиции химического индикатора, вставленного в контрольное изделие.
А.3 Стандартная контрольная упаковка
А.3.1 Стандартная контрольная упаковка используется в испытаниях с небольшой загрузкой, с полной загрузкой, испытании Бови-Дика, испытаниях детектора воздуха, влажности текстильной загрузки, а также может использоваться для продолжительных [постоянных] испытаний на соответствие требованиям, изложенным в приложении А, пунктах А.3.3, А.3.6 и А.3.7.
А.3.2 Стандартная контрольная упаковка должна составляться из чистых гладких хлопчатобумажных простыней, каждая из которых должна быть отбелена до чистого белого цвета и иметь размер порядка 900х1200 мм. Количество нитей на сантиметр в основе ткани должно быть 30, а количество уточных нитей на сантиметр - порядка 27
. Вес простыни должен составлять около 185
г/м
, а кромки, кроме боковых, должны быть подрублены.
А.3.3 Новые или загрязненные простыни должны быть выстираны, но во время стирки не должны обрабатываться никаким кондиционером для тканей. Простыни должны быть высушены, а затем подвергнуты выдержке при температуре окружающей среды от 20°С до 30°С при относительной влажности от 40% до 60%.
Примечание - Кондиционирующие агенты для тканей могут влиять на характеристики ткани и содержать добавки, способствующие образованию в стерилизаторе дополнительных неконденсирующихся газов.
А.3.4 После выдержки простыни должны быть сложены до размера примерно 220х300 мм, как показано на рисунке А.1.
А.3.5 Затем простыни должны быть сложены в стопку высотой примерно 250 мм (после нажатия на стопку рукой). Стопка простыней затем оборачивается аналогичной тканью, и обертка закрепляется стерилизационной лентой шириной не более 25 мм. Для стерилизаторов, не способных вместить больше одного модуля, высота стопки должна составлять примерно 150 мм.
А.3.6 Общий вес стандартной контрольной упаковки должен составлять примерно 7,0±0,14 кг (примерно 30 простыней), а для небольших упаковок - 4,0±0,16 кг (примерно 17 простыней). После использования стопка простыней сожмется. Если вес простыней, составляющих стопку высотой 250 мм, превысит 7,14 кг, часть простыней должна быть изъята. Аналогично, если вес простыней, составляющих стопку высотой 150 мм, превысит 4,16 кг, часть простыней также должна быть изъята.
А.3.7 Непосредственно перед использованием температура внутри стопки должна быть в пределах от 20°С до 30°С при влажности от 40% до 60%. После использования упаковка вынимается из стерилизатора и проветривается в окружающей среде с аналогичными параметрами.
А.3.8 Контрольные упаковки, состоящие из различных материалов с различными размерами, могут использоваться при условии доказательства их эквивалентности требованиям испытаний, в которых должна использоваться стандартная контрольная упаковка (см. ИСО 11140-4 [56]).
Размеры в мм
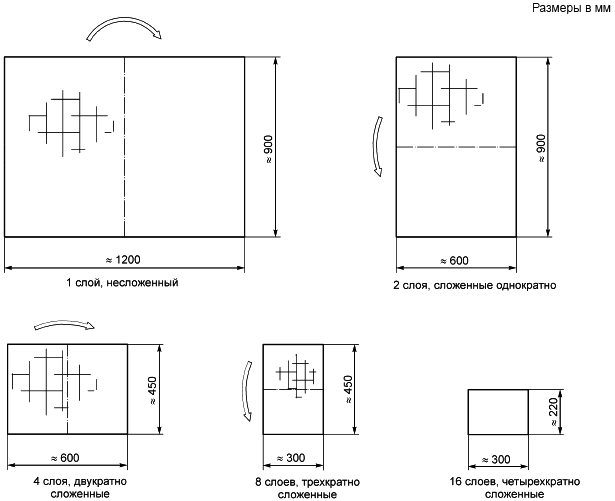
Рисунок А.1 - Складывание каждой простыни
А.4 Термометрические испытания
А.4.1 Термометрические испытания с малой загрузкой
А.4.1.1 Это испытание на проникание пара в стандартную контрольную упаковку (см. А.3). Описанную контрольную упаковку используют с целью определения полноты удаления воздуха, достаточного для аттестации стерилизационного процесса, предназначенного для обработки широкого ряда канюль, металлических и текстильных продуктов. Для выполнения данного испытания определенное количество датчиков температуры (5) размещают на разных уровнях внутри стандартной контрольной упаковки вокруг ее вертикальной оси. Рисунок 6, приведенный в EN 285, иллюстрирует принцип размещения датчиков температуры.
А.4.1.2 Критерии приемлемости для данного испытания следующие:
a) полоса температур стерилизации должна иметь нижний предел, равный температуре стерилизации, и верхний предел, равный температуре стерилизации плюс 3°С;
b) время выравнивания не должно превышать 15 с для стерилизационных камер с полезным объемом до 800 л и не превышать 30 с для камер большего размера;
c) во время периода плато температура, измеренная над контрольной упаковкой, не должна превышать температуру в контрольной точке измерения температуры в камере стерилизатора более, чем на 5°С в течение первых 60 с, и более, чем на 2°С в течение оставшегося времени;
d) во время выдержки температура, измеренная в контрольной точке измерения температуры в камере стерилизатора, любая температура, измеренная в тот же момент внутри контрольной упаковки, и соответствующая температура насыщенного пара, рассчитанная по давлению в камере, должны находиться внутри полосы температур стерилизации и не должны отличаться друг от друга более, чем на 2°С (см. пункт 6.1.2).
e) время выдержки должно быть не менее 15 мин, 10 мин и 3 мин для температур стерилизации 121°С, 126°С и 134°С соответственно.
А.4.2 Термометрические испытания с полной загрузкой
А.4.2.1 Это испытание на проникание пара в стандартную контрольную упаковку максимального размера, она дополняет испытание с малой загрузкой. Контрольная упаковка из текстиля помещается в центр полной загрузки, состоящей из текстильных изделий.
А.4.2.2 Критерии приемлемости для данного испытания такие же, как для испытания с малой загрузкой, за исключением того, что не выполняется измерение температуры над контрольной упаковкой.
А.5 Испытание Бови-Дика
А.5.1 Это испытание на проникание пара, такое же, как испытание с малой загрузкой, но предназначенное для ежедневного выполнения. В центре стандартной контрольной упаковки размещается химический индикатор, соответствующий требованиям ИСО 11140-3 [55], успешное прохождение испытания определяется по равномерному изменению цвета индикатора.
Оригинальная работа, на которой основано испытание Бови-Дика, приведена в статье Бови и др. [44].
А.5.2 Стандартная контрольная упаковка, изложенная в приложении А, подраздел А.3, представляет для процесса стерилизации нагрузку номинально такую же, как нагрузка от текстильной упаковки, описанной Бови. В качестве альтернативы для проведения испытания Бови-Дика на проникание пара со стандартной контрольной упаковкой могут использоваться индикаторы, соответствующие требованиям ИСО 11140-4 [56].
А.6 Испытание на скорость натекания воздуха
Рабочие характеристики, определенные в приложении А, подразделы А.2, А.4 и А.5, основаны на достижении низкого уровня остаточного воздуха.
Натекание воздуха в камеру стерилизатора влияет на этот уровень, и она не должна позволять давлению в камере возрастать более, чем на 0,13 кПа/мин (1,3 мБар/мин) при измерении давления в камере порядка 6 кПа (60 мБар) или ниже.
А.7 Испытания функций детектора воздуха (если имеется) с малой загрузкой, с полной загрузкой
А.7.1 Эти испытания используют для настройки детектора воздуха на регистрацию неисправности, если содержание остаточного воздуха в камере достаточно, чтобы привести к неудачному процессу с малой загрузкой (см. А.4.1) и полной загрузкой (см. А.4.2).
А.7.2 Детектор воздуха должен регистрировать неисправность, если в начале времени выравнивания наличие остаточного воздуха приведет к разнице между наиболее низкой температурой, измеренной в стандартной контрольной упаковке (используемой в испытаниях, изложенных в приложении А, подразделы А.4.1 и А.4.2), и температурой, измеренной в контрольной точке измерения температур в камере, превышающей 2°С.
Для стерилизационных камер, неспособных вместить эту контрольную упаковку из-за своих малых размеров, используется меньшая по размерам версия контрольной упаковки (смотрите А.3).
А.8 Сухость загрузки - малая и полная загрузки с текстилем, полная загрузка с металлом
Эти испытания используются для верификации того, что построение рабочего цикла, выбор параметров процесса и влажность пара таковы, что уровень остаточной влажности загрузки в конце процесса стерилизации не увеличивается более чем на 1% для текстиля, и более чем на 0,2% для металла.
А.9 Испытания на уровень звуковой мощности
Испытания, определяющие уровень звуковой мощности для использования в расчетах звуковой мощности, генерируемой стерилизатором, должны выполняться изготовителем стерилизатора в соответствии с требованиями МЭК 61010-2-040 [24].
Звуковое давление, создаваемое стерилизатором в помещении, где он установлен, должно определяться в соответствии с ИСО 3746 [58] и верифицироваться на соответствие требованиям национальных нормативов на звуковые шумы в окружающей среде.
А.10 Испытание динамического давления
Это испытание используется для верификации того, то максимальная скорость изменения давления в камере стерилизатора не приведет к повреждению упаковки изделий. Среднее изменение давления в течение любого 3-секундного интервала времени в ходе процесса стерилизации не должно превышать 1000 кПа/мин (10 Бар/мин).
А.11 Испытание качества пара
А.11.1 Неконденсируемые газы в паре влияют на удаление воздуха из камеры. Влага, содержащаяся в паре, влияет на остаточную влажность загрузки стерилизатора. Перегрев пара задерживает наличие в камере насыщенного пара. Широкий разброс давлений на линии подачи пара в стерилизатор означает недостаточную производительность источника пара, а это может повлиять на действительность значения 2°С при оценке наличия насыщенного пара (см. А.4).
Примеси в паре могут вызывать коррозию и оставлять токсичные вещества на продукте.
А.11.2 При выполнении испытаний методами, описанными в ЕН 285, к качеству подаваемого в стерилизатор пара предъявляются следующие требования:
a) количество неконденсируемых газов в паре не должно превышать 3,5% по объему;
b) минимальное значение сухости 95% (5% влажности) для загрузки стерилизатора, содержащей металл, и минимум 90% (10% влажности) для загрузки стерилизатора, содержащей текстиль;
c) максимум 25% перегрева при расширении до атмосферного давления;
d) примеси в соответствии с таблицами А.1 и А.2;
e) отклонения давления пара, не превышающие ±10% от номинального манометрического давления, измеряемого на входе в последний редукционный клапан [на линии подачи пара].
Таблица А.1 - Примеси в конденсате, полученном на входе пара в стерилизатор, рассматриваемые в аспекте коррозии материалов
Наименование | Содержание в конденсате |
Силикат ( |
|
Железо |
|
Кадмий |
|
Свинец |
|
Тяжелые металлы, за исключением железа, кадмия, свинца |
|
Хлориды ( |
|
Фосфаты ( |
|
Проводимость (при 25°С) |
|
рН (кислотность) | от 5 до 7 |
Визуально (внешние признаки) | Бесцветный, чистый, без осадка |
Жесткость ( |
|
Примечание - Метод отбора пробы конденсата изложен в ЕН 285, подраздел 22.4. | |
Таблица А.2 - Примеси в конденсате, полученные из пара, использованного в стерилизаторе, рассматриваемые в аспекте контаминации загрузки
Наименование | Содержание в конденсате из чистого пара |
Кислотность или щелочность | R |
Аммоний ( |
|
Кальций и магний |
|
Тяжелые металлы |
|
Хлориды ( |
|
Нитраты ( |
|
Сульфаты ( |
|
Окисляемые вещества |
|
Остатки после выпаривания |
|
Силикаты ( |
|
Фосфаты ( |
|
Проводимость (при 25°С) |
|
Бактериальные эндотоксины |
|
Визуально (внешние признаки) | Бесцветный, чистый, без осадка |
| |
А.12 Вода
Подаваемая вода должна иметь качество питьевой воды, а подача должна осуществляться через устройство защиты от обратного протока. Из-за влияния температуры на рабочие характеристики системы температура подаваемой воды не должна превышать 15°С. Значение жесткости воды ( щелочноземельных ионов), должна быть между 0,7 ммоль/л и 2,0 ммоль/л.
Значение жесткости, выходящее за эти пределы, может вызвать отложение накипи и проблемы с коррозией.
А.13 Сжатый воздух
Сжатый воздух должен подаваться под давлением от 600 до 800 кПа (от 5 до 7 Бар), он не должен содержать воду (в жидкой фазе), не должен содержать капель масла размером более 3 мкм, и должен быть профильтрован через фильтр 25 мкм.
А.14 Программы испытаний
Пример, показанный в таблице А.3, включает в себя испытания, необходимые для верификации достижения определенных параметров процесса, а также для оценки по полученным данным присутствия неконденсируемых газов в камере стерилизатора во время периода плато в количестве, достаточном для предотвращения проникания пара в медицинские изделия, используемые в здравоохранении.
Таблица А.3 - Пример графика испытаний для валидации и периодических испытаний
Испытание | Ссылка на ЕН 285 | Аттестация монтажа | Аттестация функциони- | Аттестация эксплуа- | Периоди- |
Испытания и проверки функций безопасности | Раздел 11 | ХХ | - | - | Х |
Качество пара (А.11): | |||||
- неконденсируемые газы; | 13.3.2, 22.1 | - | X | - | X |
- значение сухости; | 13.3.3, 22.2 | - | X | - | Х |
- перегрев; | 13.3.4, 22.3 | - | X | - | Х |
- примеси | Таблица Е.2 | - | X | - | Х |
Термометрические испытания (А.4): | |||||
- малая загрузка (А.4.1); | 8.3.1.2, 16.1 | - | XX | - | X |
- полная загрузка (А.4.2) | 8.3.1.2, 16.2 | - | XX | - | Х |
Испытание с полной загрузкой (А.2) | 8.2.5, 15 | - | XX | - | Х |
Испытание Бови-Дика (А.5) | 8.3.2, 17 | - | XX | - | X |
Скорость натекания воздуха (А.6) | 8.3.3, 18 | - | XX | - | X |
Детектор воздуха - если имеется (А.7): | |||||
- малая загрузка; | 8.3.4.2, 19.2 | - | XX | - | Х |
- полная загрузка; | 8.3.4.3, 19.3 | - | XX | - | Х |
- функции | 8.3.4.4, 19.4 | - | XX | - | Х |
Испытание на сухость загрузки (А.8): | |||||
- малая загрузка (текстиль); | 8.4.1, 20.1 | - | X | - | - |
- полная загрузка (текстиль); | 8.4.2, 20.2 | - | X | - | - |
- металл | 8.4.3, 20.3 | - | X | - | - |
Испытание динамического давления (А.10) | 10, 23 | - | - | - | Х |
Испытание продукта | - | - | - | Х | Х |
Примечание - Знак "XX" обозначает предлагаемые испытания, знак "X" - рекомендуемые испытания, "-" - необязательные испытания [испытания, которые могут не выполняться]. | |||||
| |||||
Приложение В
(справочное)
Оценка процесса стерилизации, основанная в первую очередь на биологической инактивации и сопровождающей процедуре механического удаления воздуха
В.1 Введение
В.1.1 Существует три применяемых общих метода стерилизации влажным теплом (см. ИСО 17665-1, приложения В, С и D). Знание этих трех методов позволяет пользователю принять обоснованное решение, какой метод ему применять, основываясь на знании подлежащего стерилизации продукта, наличии или отсутствии информации о микробиологической нагрузке на продукте, и понимании микробиологических рисков, возникающих в случае неудачной стерилизации.
В. 1.2 Первый метод известен как "метод биологической нагрузки", для которого идентифицируется фактическая микробиологическая нагрузка и определяется процесс с влажным теплом; при этом определении разрабатываются параметры влажного тепла, необходимые для освобождения продукта от этой специфической бионагрузки. Затем продукт испытывается на стерильность. Этот метод особенно применим в случаях, когда влияние условий стерилизации на продукт должно быть сведено к минимуму (см. ИСО 17665-1, приложение В).
В.1.3 Второй метод обычно известен как "комбинированный метод биологического индикатора и биологической нагрузки" для него идентифицируется фактическая бионагрузка, выбирается более резистентный биологический индикатор, как наиболее подходящий представитель нагрузки, и разрабатывается процесс с влажным теплом, в котором продукт может быть оценен как свободный от жизнеспособных микроорганизмов, основываясь на успешной демонстрации инактивации более устойчивых организмов, содержащихся в биоиндикаторе. Этот метод особенно применим в случаях, когда биологическая нагрузка известна и контролируема, как это бывает во многих производственных операциях (см. ИСО 17665-1, приложение С).
В.1.4 Третий метод известен как "метод полной гибели", когда фактическая микробиологическая нагрузка неизвестна или по некоторым причинам не может быть измерена, как, например, при обработке медицинских изделий многократного применения. Этот метод может также использоваться, когда подлежащие стерилизации продукты устойчивы и могут легко выдерживать условия, необходимые для достижения стерильности. При методе "полного уничтожения" выбирается микроорганизм, представительный в качестве значительной нагрузки для определенного процесса с влажным теплом. Определенный процесс (например, процесс с насыщенным паром) выражается в параметрических терминах (обычно это время и температура, объединенные с предельными значениями для пара), относящихся к кинетике летальности выбранного микроорганизма. Определяются предельные значения параметров (если это применимо), определяются также приемлемые условия летальности. Определяется приемлемый минимальный критерий (как правило, это время выдержки при заданной температуре), и к критическим параметрам применяется коэффициент безопасности с целью установления рекомендуемых условий экспозиции для продукта, подлежащего стерилизации. Для задач, в которых биологическая нагрузка не может быть определена точно, и природа и смесь продуктов могут варьироваться, обычно применяются весьма широкие границы безопасности. Этот метод особенно применим к задачам, решаемым в области здравоохранения, (см. ИСО 17665-1, приложение D).
Два примера цикла с "полной гибелью" приводятся ниже.
a) Процесс стерилизации рассчитан на обеспечение редукции микроорганизмов как минимум в 12-log, имея значение , равное одной минуте при 121°С. Валидация процесса стерилизации, базирующегося на этом значении, часто близко ассоциируется с циклом "полной гибели" с температурой 121°С, поддерживаемой в течение 15 минут.
b) Процесс стерилизации, обеспечивающий летальность в степени, превышающей требуемую для разрушения биологической нагрузки.
В.1.5 В области здравоохранения каждый стерилизатор должен периодически подтверждать свою способность выпускать обрабатываемые в нем продукты свободными от жизнеспособных микроорганизмов, таких, как на рутинно обрабатываемых в нем медицинских изделиях.
Методы, используемые для мониторинга процесса стерилизации, обеспечивают уверенность в успешном выполнении процесса в промежутках между испытаниями с биологической нагрузкой (см. таблицу В.1).
В.2 Биологическая аттестация процесса стерилизации
В.2.1 Должен быть продемонстрирован уровень обеспечения стерильности (SAL), по меньшей мере 10. Уровень обеспечения стерильности - это уровень микробиологического контроля процесса, он определяется как вероятность сохранения жизнеспособного микроорганизма на продукте, или как вероятность получения нестерильной единицы. Физические ограничения микробиологических измерений таковы, что в лучшем случае мы можем напрямую измерить уровень вероятности выживания порядка 10
. Поскольку процесс стерилизации должен базироваться на реальных данных, были разработаны стратегии, позволяющие нам разрабатывать и аттестовать процессы до уровня обеспечения стерильности 10
, используя методы косвенных измерений.
В.2.2 При необходимости стерилизации термостойких материалов обычно используется подход "полной гибели". Его простота, устойчивость и легкость валидации по сравнению с другими подходами должны сделать его подходом "по умолчанию" во всех случаях (см. ИСО 17665-1, приложение D).
В.2.3 Необходимо получение приемлемых результатов от трех последовательных циклов с походом полуцикла или полного цикла для каждого типа загрузки (см. ИСО 17665-1, приложение D, подраздел D.4).
В.2.4 Биологические индикаторы, используемые в испытаниях, должны содержать устойчивые к теплу споры, такие как споры Geobacillus stearothermophilus, и должны соответствовать требованиям соответствующих стандартов (см. ИСО 11138-1, ИСО 11138-3 и ИСО 17665-1, приложение D, подраздел D.4.1).
В.3 Биологическая нагрузка
В.3.1 Биологическая нагрузка состоит из шестнадцати свежевыстиранных 100%-ных хлопчатобумажных полотенец в хорошем состоянии. После складывания полотенца складываются друг на друга сгибами в разные стороны, формируя стопку. Один или несколько биологических индикаторов помещаются между восьмым и девятым полотенцами, примерно в геометрическом центре стопки (см. AAMI ST 79 [59]).
В.3.2 Биологическая нагрузка располагается на полке/стеллаже в горизонтальной плоскости (слои полотенец лежат горизонтально) в зоне, наименее благоприятной для стерилизации. Для стерилизаторов с гравитационным замещением воздуха испытание выполняется с полностью загруженной камерой. Для стерилизаторов с динамическим удалением воздуха, напротив, испытание выполняется с пустой камерой.
В.3.3 При использовании биологической нагрузки должен быть продемонстрирован уровень обеспечения стерильности не менее 10. Один из методов, применимых для демонстрации уровня обеспечения стерильности 10
, является метод эмпирической "полной гибели", базирующийся на 12-log снижении числа микроорганизмов при
°С в течение 1 минуты с результатом
![]() 12 при значении
12 при значении 10°С (см. работу Pflug [53]).
В.3.4 Влажность, сохраняемая тканью, не должна приводить к увеличению первичного веса (до стерилизации) биологической нагрузки более чем на 3%. На ткани не должно быть видимых влажных пятен.
В.4 Механическое удаление воздуха
В.4.1 Испытания на удаление воздуха и скорость натекания воздуха в камеру дополняют друг друга. Стерилизатор с динамическим удалением воздуха должен удовлетворять требованиям обоих испытаний.
Ни одно из этих испытаний не применимо к стерилизаторам с гравитационным замещением воздуха.
В.4.2 Эффективность работы системы удаления воздуха стерилизатора с динамическим удалением воздуха проверяется с помощью испытания на проникание пара, аналогичного испытанию Бови-Дика. (см. ИСО 11140-5).
В.4.3 Во время испытания стерилизатор с динамическим удалением воздуха должен демонстрировать среднюю скорость утечки порядка 1 мм рт.ст. в минуту или меньше за все время измерения.
В.4.4 Загрузка для испытания на проникание пара состоит из сложенных 100%-ных хлопчатобумажных хирургических полотенец, чистых и предварительно кондиционированных. Полотенца после складывания должны быть уложены друг на друга.
В.4.5 Индикаторный лист Бови-Дика помещается на центральный слой стопки. Стопка свободно (не туго) оборачивается одной двухслойной оберткой из 100%-ной хлопчатобумажной ткани (см. ИСО 11140-5).
В.4.6 Контрольная упаковка располагается горизонтально на нижней передней части загрузочного стеллажа pack рядом с дверью и над сливом камеры, при пустой камере стерилизатора.
В.4.7 Таблица В.1 демонстрирует график проведения периодических и валидационных испытаний.
Таблица В.1 - Пример графика проведения периодических и валидационных испытаний
Испытание и мониторинг | Аттестация монтажа | Аттестация функцио- | Аттестация эксплуати- | Рутинные испытания стерили- | Периоди- | Комментарий |
Испытания на соответствие типу и проверка безопасности: | X | - | - | - | - | - |
Контрольная упаковка для биологической нагрузки | - | X | - | - | X | X |
Испытание на удаление воздуха | - | X | X | X | X | |
Испытание на утечку (натекание) воздуха | - | - | - | - | - | Испытание выполняется изготовителем |
Физический мониторинг | - | X | X | X | X | - |
Мониторинг, запись данных, контроль | - | X | X | X | X | - |
Независимый датчик/самописец | - | - | - | - | - | Опционально |
Биологические индикаторы | - | X | X | X | X | - |
Химические индикаторы | - | X | X | X | X | - |
Примечание - Знак "X" обозначает рекомендуемые испытания (необходимость исполнения подлежит рассмотрению). | ||||||
Приложение С
(справочное)
Температура и давление насыщенного пара, используемого при стерилизации влажным теплом
Теоретическая температура пара (см. ИСО 17665-1, пункт 6.1.2, перечисление b) может быть определена непосредственно из приведенных ниже таблиц или рассчитана по уравнению (С.1).
Таблица С.1 - Температура и давление насыщенного пара, используемого в стерилизации влажным теплом
Давление мБар | Температура, | Давление МПа |
| Отклонение от таблиц пара |
1014,2 | 100 | 0,10142 | 99,9971 | -0,00287 |
1050,9 | 101 | 0,10509 | 100,9981 | -0,00185 |
1088,7 | 102 | 0,10887 | 101,9993 | -0,00071 |
1127,7 | 103 | 0,11277 | 103,0025 | 0,002456 |
1167,8 | 104 | 0,11678 | 104,0044 | 0,004372 |
1209 | 105 | 0,1209 | 105,0045 | 0,004526 |
1251,5 | 106 | 0,12515 | 106,0071 | 0,007106 |
1295,1 | 107 | 0,12951 | 107,0068 | 0,006789 |
1340,1 | 108 | 0,13401 | 108,0098 | 0,009813 |
1386,3 | 109 | 0,13863 | 109,0110 | 0,011046 |
1433,8 | 110 | 0,14338 | 110,0121 | 0,012132 |
1482,6 | 111 | 0,14826 | 111,0125 | 0,012517 |
1532,8 | 112 | 0,15328 | 112,0137 | 0,013662 |
1584,3 | 113 | 0,15843 | 113,0130 | 0,013041 |
1637,3 | 114 | 0,16373 | 114,0140 | 0,013976 |
1691,8 | 115 | 0,16918 | 115,0158 | 0,015808 |
1747,7 | 116 | 0,17477 | 116,0162 | 0,01617 |
1805,1 | 117 | 0,18051 | 117,0164 | 0,016372 |
1864 | 118 | 0,1864 | 118,0159 | 0,015913 |
1924,5 | 119 | 0,19245 | 119,0160 | 0,015968 |
1986,7 | 120 | 0,19867 | 120,0176 | 0,017586 |
2050,4 | 121 | 0,20504 | 121,0171 | 0,017056 |
2115,8 | 122 | 0,21158 | 122,0171 | 0,017072 |
2182,9 | 123 | 0,21829 | 123,0171 | 0,017112 |
2251,7 | 124 | 0,22517 | 124,0167 | 0,016702 |
2322,2 | 125 | 0,23222 | 125,0154 | 0,015407 |
2394,6 | 126 | 0,23946 | 126,0156 | 0,01556 |
2468,8 | 127 | 0,24688 | 127,0153 | 0,015271 |
2544,8 | 128 | 0,25448 | 128,0141 | 0,014123 |
2622,7 | 129 | 0,26227 | 129,0130 | 0,013003 |
2702,6 | 130 | 0,27026 | 130,0127 | 0,01271 |
2784,4 | 131 | 0,27844 | 131,0115 | 0,011547 |
2868,2 | 132 | 0,28682 | 132,0103 | 0,010329 |
2954,1 | 133 | 0,29541 | 133,0098 | 0,009792 |
3042 | 134 | 0,3042 | 134,0084 | 0,008352 |
3132 | 135 | 0,3132 | 135,0068 | 0,006761 |
3224,2 | 136 | 0,32242 | 136,0057 | 0,005699 |
3318,5 | 137 | 0,33185 | 137,0037 | 0,003686 |
3415,1 | 138 | 0,34151 | 138,0024 | 0,00244 |
3513,9 | 139 | 0,35139 | 139,0005 | 0,000526 |
3615 | 140 | 0,3615 | 139,9986 | -0,00142 |
| ||||
![]() , (С.1)
, (С.1)
где - теоретическая температура пара, в градусах Цельсия;
- измеренное давление, в МПа.
Число минус 273,27°С используется как значение абсолютного нуля по шкале Кельвина (0 K). Это значение используется для компенсации смещения 0,1°С между рассчитанной по таблице теоретической температурой и температурой, предписанной действующими таблицами пара.
Для получения промежуточных значений может использоваться линейная интерполяция между точками данных.
Пример расчета:
Для -0,20504 МПа и
121°С
![]() 1,584550
1,584550
![]() (-9,48654)=-1,58455+(-9,48654)=-11,071093892,7/(-11,07109)=351,6094
(-9,48654)=-1,58455+(-9,48654)=-11,071093892,7/(-11,07109)=351,6094
42,6776+351,6094=394,28705 К = 394,28705-273,27=121,0171°С
Приложение D
(справочное)
Особые соображения по процессам, используемым в области здравоохранения
D.1 Введение
Данное приложение предлагает дополнительные указания, которые могут использоваться при валидации процесса стерилизации, применяемого при обработке изделий многократного использования.
D.2 Элементы системы обеспечения качества (дополнительные указания к применению ИСО 17665-1, подраздел 4.1)
D.2.1 Руководитель с исполнительскими полномочиями должен нести ответственность за:
a) введение в действие системы обеспечения качества, пересмотры ее через регулярные промежутки времени и получение доказательств, что система понятна сотрудникам, внедрена и поддерживается с помощью текущей информации;
b) определение ролей и распределение ответственности, распределение задач и процессов, подлежащих исполнению;
c) обеспечение четкой идентификации всей цепочки отчетности;
d) обеспечение верификации и документирования изменений в процессе;
e) обеспечение составления и внедрения обзоров качества работы персонала;
f) обеспечение необходимых ресурсов для обучения персонала, наблюдения за персоналом, рабочей деятельности и аудитов системы обеспечения качества.
D.2.2 Руководитель с исполнительскими полномочиями должен понимать преимущества, создаваемые для предприятия системой обеспечения качества, в том числе финансовые, и нести ответственность за:
a) назначение персонала, обученного выполнению аудитов качества и внедрение инициатив, способствующих повышению качества;
b) определение и распределение ответственности между оперативным и управленческим персоналом;
c) определение квалификации, компетенций и ответственности каждого ответственного лица, назначенного для выполнения специфической задачи (задач);
d) обеспечение необходимой квалификации, уровня образования и производственного обучения персонала;
e) внедрение мероприятий по предотвращению инфекций и программы контроля, включая процедуры и протоколы;
f) принятие мер по охране здоровья и безопасности персонала;
g) определение процедур для субпоставщиков, включая работающих как вне, так и внутри учреждения здравоохранения (если применимо);
h) определение процедур, обеспечивающих непрерывное поддержание качества и компетенции персонала;
i) обеспечение наличия действующих процедур контроля и мониторинга всех фаз работы, наличие документации, обеспечивающей выполнение всех требований стандартов, руководств и нормативных актов, относящихся к хранению, использованию чистящих и дезинфицирующих агентов, и за их использование персоналом с соблюдением всех указаний, имеющихся на этикетках;
j) обеспечение соответствия приобретенных стерилизаторов законодательным требованиям и их спецификациям (см. ИСО 17665-1, подраздел 6.2);
k) обеспечение правильности монтажа стерилизаторов с соблюдением их правильного функционирования, безопасности персонала и охраны окружающей среды;
I) обеспечение соответствия характеристик питающих стерилизаторы сред требованиям изготовителя стерилизатора;
m) обеспечение валидации вновь приобретенных стерилизаторов до их ввода в эксплуатацию по документированной схеме, включая аттестацию монтажа, аттестацию функционирования и аттестацию эксплуатируемого оборудования;
n) обеспечение выполнения по документированным схемам периодических испытаний стерилизаторов на ежегодной, ежеквартальной, еженедельной и - в некоторых случаях - ежедневной основе;
о) обеспечение наличия договоров на техническое обслуживание с внешними организациями, имеющими полностью обученный и допущенный к работам персонал, или программ технических осмотров и обслуживания собственным техническим персоналом, полностью обученным и допущенным к таким работам;
р) обеспечение наличия и исполнения документированных процедур производства, контроля качества и охраны труда, соответствующих требованиям законодательства и лучших принятых производственных практик;
q) обеспечение наличия и исполнения процедур, связанных с неисправностями, несчастными случаями и опасными ситуациями;
r) обеспечение документированного исполнения рекомендаций изготовителя оборудования по регулярному техническому обслуживанию и периодическим техническим осмотрам (смотрите ИСО 17665-1, раздел 12 и подраздел 12.3);
s) обеспечение периодической калибровки, осмотра, проверки и техобслуживания всех измерительных цепей, встроенных в стерилизатор или используемых в работе со стерилизатором; калибровка средств измерения должна выполняться с помощью контрольно-измерительного оборудования, калиброванного и прослеживаемого до национальных стандартов;
t) обеспечение предоставления изготовителем стерилизатора информации/графиков относительно любых частей и компонентов, требующих рутинной замены, и обеспечение доступности этой информации для пользователя.
D.2.3 Руководитель с исполнительскими полномочиями должен уделять внимание таким эксплуатационным аспектам, как:
a) организация и обустройство рабочих зон (зоны деконтаминации, приготовления, стерилизации и хранения стерильной продукции), контроль окружающей среды, санитарные комнаты с умывальниками для мытья рук, рабочие поверхности, контроль потоков персонала и транспортных средств, средства персональной защиты для сотрудников, соблюдение установленной формы одежды персоналом;
b) подготовка медицинских изделий;
c) очистка и дезинфекция;
d) сборка и проверка функционирования сложных изделий;
e) упаковка;
f) загрузка стерилизаторов, их эксплуатация, разгрузка и выпуск продукта;
g) хранение и распределение;
h) необходимость наличия и наличие системы, обеспечивающей прослеживание каждого медицинского изделия;
i) обеспечение и поддержание стерильности;
j) соглашения о закупках (договоры);
k) обеспечение технического обслуживания и поддержание качества работы стерилизаторов;
I) управление и отчетность об инцидентах, требующих внимания и/или действий;
m) обеспечение стерилизации медицинских изделий в соответствии с требованиями их семейств продуктов и/или рекомендациями их изготовителей;
n) обеспечение стерилизации медицинских изделий, трудно поддающихся очистке или стерилизации.
D.2.4 Руководитель с исполнительскими полномочиями должен уделять внимание требованиям к документированию процессов стерилизации и обеспечить включение в эту документацию таких аспектов, как:
a) регулярные аудиты качества системы документирования;
b) обеспечение доступности данных персоналу;
c) стандартные рабочие процедуры, имеющие достаточную информацию обо всех критических ступенях процесса стерилизации;
d) руководства, диаграммы и визуальные указатели, которые должны быть всегда доступны персоналу;
е) все отчеты аудита процессов и оборудования;
f) все изменения, внесенные в процессы; изменения должны документироваться, утверждаться и своевременно доводиться до соответствующего персонала.
D.3 Описание процесса и оборудования (дополнительные указания к ИСО 17665-1, раздел 6)
D.3.1 Процесс (дополнительные указания к ИСО 17665-1, подраздел 6.1)
D.3.1.1 Два подхода к оценке процесса стерилизации изложены в приложениях А и В.
D.3.1.2 Если наличие условий стерилизации на медицинском изделии (изделиях) не может быть спрогнозировано на основании испытаний, описанных в приложении А, может оказаться необходимым выполнение испытаний, предписанных приложениями А и В.
D.3.2 Оборудование (дополнительные указания к ИСО 17665-1, подраздел 6.2)
Стерилизаторы, предназначенные для использования в сфере здравоохранения, должны быть снабжены процессом (процессами) стерилизации, рассчитанным на стерилизацию широкого ряда медицинских изделий, рутинно используемых в учреждениях здравоохранения.
D.4 Определение продукта в учреждениях здравоохранения (дополнительные указания к ИСО 17665-1, раздел 7)
D.4.1 Дополнительные указания к ИСО 17665-1, подраздел 7.1
Маловероятно, чтобы учреждение здравоохранения участвовало в разработке и конструировании медицинского изделия. Более вероятно то, что учреждение здравоохранения может оказаться перед выбором приобретения одного или нескольких коммерческих продуктов. В определенных обстоятельствах учреждение здравоохранения все же может быть привлечено к разработке и конструированию нового или к модификации старого медицинского изделия. В таких обстоятельствах семейство продукта должно быть определено, и требования ИСО 17665-1 подлежат выполнению.
D.4.2 Дополнительные указания к ИСО 17665-1, подраздел 7.3
Учреждения здравоохранения зачастую укладывают несколько различных коммерческих продуктов в одну упаковку. Для такой комбинации следует определять семейство продукта. В большинстве случаев определенное семейство продукта должно совпасть с семейством продукта того медицинского изделия в упаковке, которое известно как наиболее трудно поддающееся стерилизации.
D.4.3 Дополнительные указания к ИСО 17665-1, подраздел 7.10
Использование изделий, трудно поддающихся очистке или разборке, должно тщательно рассматриваться перед их приобретением, поскольку успешно стерилизовать можно только хорошо очищенное изделие. Инструкции по повторной обработке должны предоставляться изготовителем медицинского изделия. Для инструментов, изготовленных до выпуска ИСО 17664 [23], следует выполнять требования региональных или национальных нормативных документов.
D.5 Определение процесса (дополнительные указания к ИСО 17665-1, раздел 8)
D.5.1 Дополнительные указания к ИСО 17665-1, подраздел 8.1
D.5.1.1 Для повторной обработки изделий в учреждениях здравоохранения применяется процесс стерилизации, основанный на рекомендациях национальных и региональных фармакопей по температуре стерилизации и времени выдержки, и/или разработанный исходя из параметров процесса, специфицированных изготовителем медицинского изделия и/или стерилизатора.
D.5.1.2 Уже разработан ряд стандартов для стерилизаторов, пригодных для обработки широкого ряда медицинских изделий. Изготовителем стерилизатора может быть рекомендован процесс стерилизации, удовлетворяющий требованиям, описанным в соответствующем стандарте.
D.5.1.3 Может использоваться и несхожий, но уже существующий процесс стерилизации, который ранее был определен для обработки семейства продукта, к которому отнесено новое медицинское изделие, при условии, что размер, конструкция и конструкционные материалы нового медицинского изделия укладываются в соответствующие характеристики семейства продукта.
D.5.1.4 Учреждения здравоохранения, довольно часто, укладывают несколько различных коммерческих продуктов в одну упаковку. В таких случаях учреждение должно учитывать требования инструкций по повторной обработке, выпущенных для каждого изделия, укладываемого в общую упаковку, и учитывать требования к семействам изделий, к которым отнесены эти продукты. Учреждение здравоохранения должно рассмотреть рекомендации отдельных поставщиков коммерческих продуктов по повторной обработке и сравнить их с условиями существующего в учреждении процесса (процессов) стерилизации. В результате может оказаться возможным использовать существующий процесс, но может понадобиться и разработка нового процесса стерилизации, основанного на рекомендациях изготовителя медицинского изделия. Любое отклонение от предлагаемого процесса стерилизации должно быть согласовано с изготовителем медицинского изделия или валидировано ответственным лицом (см. ИСО 17665-1, приложение D, пункт D.8.3.1 и приложение А, пункт А.4.2).
D.5.1.5 Если планируемый к использованию стерилизационный процесс не рекомендован изготовителем медицинского изделия и не был ранее определен как пригодный к обработке семейства продукта, к которому отнесено новое изделие, то должно быть проведено детальное сравнение нового продукта с продуктами, которые изначально были определены для обработки этим процессом. Сравнение должно включать в себя физические характеристики наряду с тепловой и биологической оценкой, где это применимо.
D.5.2 Дополнительные указания к ИСО 17665-1, подраздел 8.3
Для каждого медицинского изделия должны учитываться физические параметры и ограничения в части экспозиции, идентифицированные изготовителем медицинского изделия. Несоблюдение инструкций изготовителя изделия по его повторной обработке может пагубно отразиться на рабочих характеристиках изделия и привести к прекращению действия гарантийных обязательств изготовителя.
D.5.3 Дополнительные указания к ИСО 17665-1, подраздел 8.4
Для многих медицинских изделий удаление воздуха является критическим фактором при прогнозировании наличия насыщенного пара в местах, трудно поддающихся стерилизации. Соответствующие испытания и критерии приемлемости для факторов, способных повлиять на проникание пара, приведены в приложениях А и В.
D.5.4 Дополнительные указания к ИСО 17665-1, подраздел 8.12
Процесс стерилизации, основывающийся на биологической нагрузке, в большинстве случаев непрактичен и не пригоден для обработки медицинских изделий многократного использования.
D.6 Валидация (дополнительные указания к ИСО 17665-1, раздел 9)
D.6.1 Аттестация функционирования (дополнительные указания к ИСО 17665-1, пункт 9.3.1)
D.6.1.1 При монтаже любого нового стерилизатора, модификации имеющегося оборудования с целью выполнения им нового процесса стерилизации или при изменении питающих сред, изготовитель стерилизатора или сторона, несущая ответственность за процесс стерилизации, должны определить семейство (семейства) продукта, которые могут обрабатываться в стерилизаторе, а также требования к рабочим характеристикам и испытания, которые должны использоваться для верификации эффективности процесса стерилизации. Изменения в процессе стерилизации, способные повлиять на его эффективность, включают в себя изменения параметров процесса.
D.6.1.2 Требования к рабочим характеристикам и испытания, которые могут потребовать утверждения:
а) эффективное удаление воздуха, полученное в ходе выполнения процесса: оно может быть спрогнозировано по рабочему циклу; воспроизводимость может быть нарушена за счет натекания воздуха в камеру, наличия в паре неконденсируемых газов и скорости изменения температуры в камере и в загрузке стерилизатора;
b) примеси [контаминанты], осевшие на медицинском изделии: это может быть спрогнозировано по примесям, содержащимся в паре;
c) проникание пара в те части медицинского изделия, из которых затруднено удаление воздуха: это может быть спрогнозировано путем сравнения температуры, измеренной в контрольном устройстве, с температурой, измеренной в контрольной точке измерения температуры в камере; контрольное устройство должно представлять собой такую же нагрузку, как и представляемое им медицинское изделие (изделия), а метод должен быть верифицирован с помощью химических или биологических индикаторов, помещаемых в контрольное изделие или в медицинские изделия;
d) сухость загрузки для упакованных изделий: это может быть определено как визуальным осмотром, так и по изменению массы загрузки.
D.6.1.3 Если существующий процесс стерилизации должен использоваться для обработки нового медицинского изделия и/или новой конфигурации загрузки, соответствие требованиям к рабочим характеристикам, установленным в ходе первичной или последующей аттестации функционирования должно быть подтверждено до выполнения аттестации эксплуатируемого оборудования. Это может быть сделано путем анализа данных, полученных из рутинной обработки, и/или по результатам периодических испытаний, или путем повторения аттестации функционирования.
D.6.2 Аттестация эксплуатируемого оборудования (дополнительные указания к ИСО 17665-1, подраздел 9.4)
D.6.2.1 Дополнительные указания к ИСО 17665-1, пункт 9.4.1
D.6.2.1.1 Эффективность и воспроизводимость процесса стерилизации и его параметров должны быть известны для полного ряда семейств продуктов и конфигураций загрузки, установленных для данного процесса стерилизации.
D.6.2.1.2 Из ряда медицинских изделий, семейств продуктов и конфигураций загрузки, которые могут обрабатываться процессом стерилизации, должна быть выбрана и идентифицирована загрузка, наиболее трудно поддающаяся стерилизации. Рекомендации по признакам, характеризующим трудность стерилизации, должны быть даны изготовителем медицинского изделия и/или изготовителем стерилизатора.
D.6.2.1.3 Аттестация эксплуатируемого оборудования должна выполняться с этой загрузкой, и при успешном завершении она может быть признана действительной для остальных возможных комбинаций из этого ряда.
D.6.2.1.4 Если на медицинском изделии после завершения процесса оказывается влага, может понадобиться его особая ориентация и/или место размещения в камере.
D.6.2.1.5 Некоторые медицинские изделия могут нуждаться в предварительной обработке, например, в уравнивании их температуры с температурой окружающей среды или с иной специфической температурой и влажностью.
D.6.2.2 Дополнительные указания к ИСО 17665-1, пункт 9.4.2
Количество используемых датчиков должно обеспечивать запись достаточного объема данных, необходимых для демонстрации эффективности процесса. Опытным путем установлено, что для типовой для здравоохранения загрузки и типового полезного объема камеры (порядка 400 л) может быть достаточно от 5 до 12 датчиков температуры.
D.6.2.3 Дополнительные указания к ИСО 17665-1, пункт 9.4.4
Данные, необходимые для определения пределов процессных параметров и критериев приемлемости для периодических испытаний должны определяться на основе данных, полученных в ходе испытаний аттестации эксплуатируемого оборудования. Сюда должны входить термометрические данные и результирующие данные химических и/или биологических индикаторов, размещенных в труднодоступных местах. Следует также оценивать возможность получения ложных термометрических данных от измерения температуры воздуха вместо пара.
D.6.3 Рассмотрение и утверждение результатов валидации (дополнительные указания к ИСО 17665-1, пункт 9.5.2)
Должны быть представлены данные, подтверждающие, что:
a) для каждого испытания, сделанного в ходе аттестации функционирования и аттестации эксплуатируемого оборудования, имеется его спецификация, и что в каждом испытании были достигнуты критерии приемлемости;
b) если испытание (испытания) должно выполняться периодически с целью верификации сохранения эффективности процесса стерилизации, в рамках его спецификации, то данное испытание и требования к его характеристикам являются производными от данных, полученных в ходе аттестации функционирования, аттестации эксплуатируемого оборудования и переаттестации, если это необходимо;
c) было идентифицировано семейство продукта, представляющее собой "худший случай" для загрузки стерилизатора;
d) если было идентифицировано любое ограничение по способу или месту размещения изделия в загрузке, то это ограничение отражено в описании конфигурации загрузки.
D.7 Текущий мониторинг и контроль (дополнительные указания к ИСО 17665-1, раздел 10)
D.7.1 Дополнительные указания к ИСО 17665-1, подраздел 10.1
D.7.1.1 Действия по текущему мониторингу и контролю могут быть разделены по периодичности [частоте] их выполнения. В число этих действий по возможности должны входить:
а) проверка скорости натекания воздуха в камеру для установления уровня этого натекания в камере стерилизатора;
b) проверка результатов исполнения планового периодического и внепланового текущего технического обслуживания;
c) испытание на проникание пара для оценки влияния остаточного воздуха и неконденсируемых газов на эффективность процесса стерилизации;
d) проверка вхождения загрузки/семейств продуктов, подлежащих стерилизации, в отчеты об аттестации/ переаттестации эксплуатируемого оборудования;
e) проверка нахождения параметров процесса в рамках их специфицированных допусков в ходе выполнения процесса стерилизации;
f) проверка способности срабатывания химических и/или биологических индикаторов согласно их спецификациям;
g) проверка способности устройств контроля процесса, детекторов воздуха или систем мониторинга процесса срабатывать согласно их спецификациям;
h) проверка (визуальная) целостности упаковок, их сухости, правильности маркировки и срабатывания химических индикаторов на упаковках после выгрузки из камеры стерилизатора.
D.7.1.2 Загрузка стерилизатора обычно состоит из медицинских изделий, относящихся к различным семействам продуктов, при этом в разных циклах стерилизации этот состав изделий может меняться. Объем остаточного неконденсируемого газа, косвенно определяемый периодическими испытаниями, должен быть таким, чтобы в определенных пределах обеспечивалось немедленное проникание пара с загрузку стерилизатора, имеющую сопротивление прониканию, равное или меньшее, чем сопротивление загрузки, признанной "худшим случаем". Основываясь на таком уровне остаточного воздуха, допускаются произвольные конфигурации загрузки, а любые ограничения обычно относятся к остаточной влажности загрузки. Для одного семейства продуктов и конфигурации загрузки может быть обнаружена воспроизводимость в небольшом коридоре допусков, и она должна быть установлена в ходе аттестации эксплуатируемого оборудования. Медицинское изделие всегда должно ассоциироваться с определенным семейством продуктов, включенным в список изделий, обрабатываемых данным процессом. Семейство продуктов может состоять как из одного медицинского изделия, так и из группы схожих медицинских изделий.
D.7.2 Дополнительные указания к ИСО 17665-1, подраздел 10.3
Очень важно, чтобы любое изменение, способное повлиять на количество остаточного воздуха в камере, было идентифицировано с помощью таких средств, как устройство контроля процесса и/или воздушный детектор и испытание на проникание пара.
D.8 Выпуск продукта после стерилизации (дополнительные указания к ИСО 17665-1, подраздел 11.1)
D.8.1 Должны иметься процедуры аудита оборудования, продукта и процесса стерилизации. Ниже приведен пример:
Стерилизатор должен удовлетворять требованиям к его назначению, и это обстоятельство должно быть отмечено в документации по выпуску продукта. Эта декларация должна основываться на следующих фактах:
a) успешно выполняемое плановое техническое обслуживание;
b) соответствие требованиям к рабочим характеристикам по условиям периодических и текущих испытаний;
c) отсутствие изменений в системе генерации/подачи пара и других рабочих сред;
d) соответствие требованиям к рабочим характеристикам для рутинной обработки по данным из последних выполненных производственных циклов.
D.8.2 Успешно завершенный процесс стерилизации включает в себя:
a) соответствие требованиям к охране окружающей среды в зонах подготовки, упаковки и стерилизации;
b) соответствие требованиям к гигиене персонала и контролю;
c) соответствие процедурам подготовки, упаковки и маркировки медицинских изделий;
d) соотнесение каждого медицинского изделия или группы изделий к определенной категории (категориям) продукта;
е) каждая загрузка стерилизатора валидирована для процесса стерилизации, предназначенного для ее обработки;
Примечание - При условии, что загрузка, представляющая собой наибольшую нагрузку на процесс стерилизации, успешно прошла аттестацию эксплуатируемого оборудования, другие загрузки с меньшей нагрузкой на процесс могут быть классифицированы как валидированные.
f) достижение заданных температуры стерилизации и длительности периода плато;
g) регистрация всего рабочего цикла и его номера;
h) специфицированная реакция химического индикатора, если он использовался;
i) подтверждение, что при визуальном осмотре каждая выгруженная упаковка была сухой и не имела повреждений.
D.8.3 Маркировки на каждой упаковке и (если применимо) на каждом медицинском изделии должны позволять идентификацию:
a) использованного для обработки стерилизатора;
b) номера цикла стерилизационного процесса;
c) самой упаковки;
d) того, что упаковка/изделие "прошло обработку".
D.8.4 Неудачное завершение периодического испытания, в том числе ежедневного теста Бови-Дика, если это применимо, должно приводить к выводу стерилизатора из эксплуатации. Если были использованы биологические индикаторы, обнаружение жизнеспособных микроорганизмов должно приводить к перемещению продуктов в карантин на время проведения расследования причин. Расследование может заканчиваться решением об уничтожении карантинной партии продукта и/или об отзыве уже выпущенной партии продукта.
D.9 Поддержание эффективности процесса (дополнительные указания к ИСО 17665-1, раздел 12)
D.9.1 Повторная калибровка (дополнительные указания к ИСО 17665-1, подраздел 12.2)
Верификация калибровки [поверка] каждой измерительной цепи должна выполняться:
a) ежегодно;
b) после внепланового технического обслуживания или текущего ремонта измерительной цепи;
c) при получении доказательства неточности измерений, выполняемых цепью.
D.9.2 Техническое обслуживание оборудования (дополнительные указания к ИСО 17665-1, пункт 12.3.1)
Техническое обслуживание или внесение изменений в питающие среды (например, в централизованный парогенератор) должны доводиться до сведения конечного пользователя, поскольку, например, изменение методов обработки воды, подаваемой в бойлер, может изменить уровни содержания в паре неконденсируемых газов, влаги и/или химических примесей до превышения ими допустимых пределов. Такие изменения подлежат оценке в части их способности к созданию несоответствий со спецификациями процесса, и при необходимости должны выполняться и документироваться корректирующие действия.
D.9.3 Повторная аттестация (переаттестация) (дополнительные указания к ИСО 17665-1, пункт 12.4.1)
Если применимо, рутинная переаттестация функционирования и переаттестация эксплуатируемого оборудования должны в общем случае выполняться ежегодно (примеры смотрите в таблице А.3). Переаттестация функционирования должна повторять некоторые или все испытания, предусмотренные для этого вида аттестации. Переаттестация эксплуатируемого оборудования должна повторять исследования по меньшей одной из рутинно обрабатываемых стерилизационных загрузок, для которой сохранены данные из предыдущей аттестации эксплуатируемого оборудования. Если полученные значения остаются в тех же пределах, что и в предыдущем исследовании, воспроизводимость процесса должна быть признана приемлемой.
Приложение Е
(справочное)
Указатель нормативных статей и параграфов ИСО 17665-1 и цитируемых ссылок или соответствующих указаний, имеющихся в ИСО 17665-1 и в документе ИСО/ТС 17665-2
Примечание - Данная таблица содержит указатель ссылок [на цитируемые документы], содержащихся в разделах и пунктах ИСО 17665-1, и соответствующих указаний и разъяснений, приведенных в приложении А к ИСО 17665-1 или в основном тексте данной технической спецификации, а также в особых соображениях относительно учреждений здравоохранения, приведенных в приложении D к данной технической спецификации.
Нормативный раздел/пункт в ИСО 17665-1:2006 | Цитируемые ссылки в ИСО 17665-1:2006 | Соответствующие указания и разъяснения к ИСО 17665-1:2006, приложение А | Соответствующие указания и разъяснения в основном тексте ИСО/TS 17665-2 (разделы 1-12) | Особые соображения и указания относительно учреждений здравоохранения в ИСО/TS 17665-2, приложение D |
4 Системы управления качеством | - | А.1.2.4 | - | - |
4.1 Документирование | Применимые разделы и пункты ИСО 13485 | А.4.1 | - | - |
4.2 Ответственность руководства | - | А.4.2 | - | D.2 |
4.3 Реализация продукта | Применимые разделы и пункты ИСО 13485 | А.4.3 | - | - |
4.4 Измерения, анализ и улучшение - контроль продукта, не соответствующего требованиям | Применимые пункты ИСО 13485
| А.4.4 | - | - |
5 Описание стерилизующего агента | - | А.5 | 5 | - |
5.1 Стерилизующий агент | - | 5.1 | - | |
5.2 Микробоцидная эффективность | - | 5.2 | - | |
5.3 Воздействие на материал | - | 5.3 | - | |
5.4 Экологические факторы | - | 5.4 | - | |
6 Описание процесса и оборудования | - | А. 6 | 6 | - |
6.1 Процесс | - | 6.1 | D.3.1 | |
6.1.1 Общее | - | 6.1.1 | ||
6.1.2 Процессы с насыщенным паром | - | 6.1.2 | ||
6.1.3 Процессы с упакованными продуктами | - | А. 6 | 6.1.3 | D.3.1 |
6.2 Оборудование | - | 6.2 | D.3.2 | |
7 Определение продукта | ИСО 11607-1 | А.7 | 7 | D.4 |
8 Определение процесса | Применимые части ИСО 11140 ИСО 11737-1 | А.8 | 8 | D.5 |
9 Валидация | - | А.9 | 9 | D.6 |
9.1 Общее | - | 9.1 | - | |
9.2 Аттестация монтажа (IQ) | - | 9.2 | - | |
9.2.1 Оборудование | - | 9.2.1 | - | |
9.2.2 Установка | - | 9.2.2 | - | |
9.2.3 Работа | - | 9.2.3 | - | |
9.3 Аттестация функционирования (OQ) | - | 9.3 | D.6.1 | |
9.4 Аттестация эксплуатируемого оборудования(PQ) | ИСО 17665-1:2006, приложения В, С и D ЕН 285 | 9.4 | D.6.2 | |
9.5 Анализ и утверждение валидации | - | 9.5 | D.6.3 | |
10 Текущий мониторинг и контроль | - | А. 10 | 10 | D.7 |
11 Выпуск продукции после стерилизации | - | А. 11 | 11 | D.8 |
12 Поддержание эффективности процесса | - | А.12 | 12 | D.9 |
12.1 Демонстрация непрерывной эффективности | - | 12.1 | - | |
12.2 Повторная калибровка | - | 12.2 | D.9.1 | |
12.3 Техническое обслуживание оборудования | - | 12.3 | D.9.2 | |
12.4 Повторная оценка | - | 12.4 | D.9.3 | |
12.5 Оценка изменений | - | - | - |
Приложение ДА
(справочное)
Сведения о соответствии ссылочных международных стандартов национальным стандартам Российской Федерации
Таблица ДА.1
Обозначение ссылочного международного стандарта | Степень соответствия | Обозначение и наименование соответствующего национального стандарта |
ИСО 17665-1 | IDT | ГОСТ Р ИСО 17665-1-2016 "Стерилизация медицинской продукции. Влажное тепло. Часть 1. Требования к разработке, валидации и текущему контролю процесса стерилизации медицинских изделий" |
Примечание - В настоящей таблице использовано следующее условное обозначение степени соответствия стандарта: | ||
Библиография
[1] | ИСО 10993-1 | Оценка биологическая медицинских изделий. Часть 1. Оценка и испытания в рамках процесса управления рисками (Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management system) |
[2] | ИСО 10993-17 | Оценка биологическая медицинских изделий. Часть 17. Установление допустимых пределов для выщелачиваемых веществ (Biological evaluation of medical devices - Part 17: Establishment of allowable limits for leachable substances) |
[3] | ИСО 11138-1:2006 | Стерилизация медицинской продукции. Биологические индикаторы. Часть 1. Общие требования (Sterilization of health care products - Biological indicators - Part 1: General Requirements) |
[4] | ИСО 11138-3:2006 | Стерилизация медицинской продукции. Биологические индикаторы. Часть 3. Биологические индикаторы для методов стерилизации паром (Sterilization of health care products - Biological indicators - Part 3: Biological indicators for moist heat sterilization processes) |
[5] | ИСО/TC 11139:2006 | Стерилизация продуктов в здравоохранении - Словарь (Sterilization of healthcare products - Vocabulary) |
[6] | ISO 11737-1:2006 | Стерилизация медицинских изделий. Микробиологические методы. Часть 1. Оценка популяции микроорганизмов на продукции (Sterilization of medical devices - Microbiological methods - Part 1: Determination of a population of microorganisms on products) |
[7] | ИСО 11737-2:2009 | Стерилизация медицинских изделий. Микробиологические методы. Часть 2. Испытания на стерильность, проводимые при валидации процессов стерилизации (Sterilization of medical devices - Microbiological methods - Part 2: Tests of sterility performed in the definition, validation and maintenance of a sterilization process) |
[8] | ИСО 11607-1:2006 | Упаковка медицинских изделий, стерилизуемых на завершающей стадии производства. Часть 1. Требования к материалам, барьерным системам стерилизации и системам упаковки (Packaging for terminally sterilized medical devices - Part 1: Requirements for materials, sterile barrier systems and packaging systems) |
[9] | ИСО 11607-2:2006 | Упаковка медицинских изделий, стерилизуемых на завершающей стадии производства. Часть 2. Требования к валидации процессов формирования, герметизации и сборки (Packaging for terminally sterilized medical devices - Part 2: Validation requirements for forming, sealing and assembly processes) |
[10] | ИСО 13485:2003 | Медицинские изделия - Системы менеджмента качества - Требования к нормативным целям (Medical devices - Quality management systems - Requirements for regulatory Purposes) |
[11] | ИСО 14001 | Системы экологического менеджмента. Требования и руководство по применению (Environmental management systems - Requirements with guidance for use) |
[12] | ИСО 14040 | Экологический менеджмент. Оценка жизненного цикла. Принципы и структура (Environmental management - Life cycle assessment - Principles and framework) |
[13] | ИСО 14161:2009 | Стерилизация медицинской продукции. Биологические индикаторы. Руководство по выбору, использованию и интерпретации результатов (Sterilization of health care products - Biological indicators - Guidance for the selection, use and interpretation of results) |
[14] | ИСО 14644-1 | Помещения чистые и связанные с ними контролируемые среды. Часть 1. Классификация чистоты воздуха (Cleanrooms and associated controlled environments - Part 1: Classification of air cleanliness) |
[15] | ИСО 14937:2009 | Стерилизация медицинской продукции. Общие требования к определению характеристик стерилизующего средства и разработке, валидации и текущему контролю процесса стерилизации медицинских изделий (Sterilization of health care products - General requirements for characterization of a sterilizing agent and the development, validation and routine control of a sterilization process) |
[16] | ИСО/ТР 14969:2004 | Медицинские изделия - Системы менеджмента качества - Руководство по применению стандарта ISO 13485: 2003 (Medical devices - Quality management systems - Guidance on the application of ISO 13485: 2003) |
[17] | ИСО 14971 | Изделия медицинские. Применение менеджмента риска к медицинским изделиям (Medical devices - Application of risk management to medical devices) |
[18] | ИСО 15882:2008 | Стерилизация медицинской продукции. Химические индикаторы. Руководящие указания по отбору, применению и интерпретации результатов (Sterilization of health care products - Chemical indicators - Guidance for selection, use and interpretation of results) |
[19] | ИСО 15883-1 | Аппараты для мойки-дезинфекции. Часть 1. Общие требования, термины, определения и испытания (Washer-disinfectors - Part 1: General requirements, terms and definitions and tests) |
[20] | ИСО 15883-2 | Аппараты для мойки-дезинфекции. Часть 2. Требования и методы испытаний аппаратов, использующих термическую дезинфекцию хирургических инструментов, оборудования для анестезии, поильников, чашек, приемников, посуды, изделий из стекла и т.д. (Washer-disinfectors - Part 2: Requirements and tests for washer-disinfectors employing thermal disinfection for surgical instruments, anaesthetic equipment, bowls, dishes, receivers, utensils, glassware, etc) |
[21] | ИСО 15883-4 | Моюще-дезинфицирующие машины. Часть 4. Требования и испытания моюще-дезинфицирующих машин с химической дезинфекцией для термолабильных эндоскопов (Washer-disinfectors - Part 4: Requirements and tests for washer-disinfectors employing chemical disinfection for thermolabile endoscopes) |
[22] | ИСО 15883-5 | Аппараты для мойки-дезинфекции. Часть 5. Загрязнения для проведения испытания и методы, демонстрирующие эффективность мойки (Washer-disinfectors - Part 5: Test soils and methods for demonstrating cleaning Efficacy) |
[23] | ИСО 17664:2004 | Стерилизация медицинских изделий. Информация, предоставляемая изготовителем для обработки повторно стерилизуемых медицинских изделий (Sterilization of medical devices - Information to be provided by the manufacturer for the processing of resterilizable devices) |
[24] | МЭК 61010-2-040 | Безопасность электрических контрольно-измерительных приборов и лабораторного оборудования. Часть 2-040. Дополнительные требования к стерилизаторам и моечным дезинфекторам, применяемым для обработки медицинских материалов (Safety requirements for electrical equipment for measurement, control and laboratory use - Part 2-040: Particular requirements for sterilizers and washer-disinfectors used to treat medical materials) |
[25] | ЕН 285 | Стерилизация. Паровые стерилизаторы. Большие стерилизаторы (Sterilization - Steam sterilizers - Large sterilizers) |
[26] | ЕН 867-5 | Системы небиологические для использования в стерилизаторах. Часть 5. Требования к индикаторным системам и испытуемым образцам для испытания характеристик малых стерилизаторов типа В и типа С. (Non-biological systems for use in sterilizers - Part 5: Specification for indicator systems and process challenge devices for use in performance testing for small sterilizers Type В and Type S) |
[27] | ЕН 868-2:1999 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 2. Упаковка для стерилизации. Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 2: Sterilization wrap - Requirements and test methods) |
[28] | EN 868-3:1999 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 3. Бумага для изготовления бумажных пакетов (технические условия в EN 868-4) и изготовления прозрачных пакетов и трубок (технические условия в EN 868-5). Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 3: Paper for use in the manufacture of paper bags (specified in EN 868-4) and in the manufacture of pouches and reels (specified in EN 868-5) - Requirements and test methods) |
[29] | EN 868-4:1999 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 4. Бумажные пакеты. Требования и методы испытания (Packaging materials and systems for medical devices which are to be sterilized - Part 4: Paper bags - Requirements and test methods) |
[30] | EH 868-5:1999 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 5. Термостойкие самозапечатывающиеся пакеты и трубки из бумаги и пластиковой пленки. Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 5: Heat and self-sealable pouches and reels of paper and plastic film construction - Requirements and test methods) |
[31] | EH 868-8:1999 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 8. Стерилизационные контейнеры, пригодные для повторного использования, для паровых стерилизаторов по EN 285. Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 8: Re-usable sterilization containers for steam sterilizers conforming to EN 285 - Requirements and test methods) |
[32] | EH 868-9:2000 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 9. Нетканые материалы из полиолефинов без покрытия. Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 9: Uncoated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches, reels and lids - Requirements and test methods) |
[33] | EH 868-10:2000 | Материалы и системы для упаковки медицинских приборов, предназначенных для стерилизации. Часть 10. Нетканые материалы из полиолефинов с клеевым покрытием. Требования и методы испытаний (Packaging materials and systems for medical devices which are to be sterilized - Part 10: Adhesive coated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches, reels and lids - Requirements and test methods) |
[34] | БС ЕН 12442-1:2000 | Ткани животных и их производные, применяемые для изготовления медицинских изделий. Часть 1. Анализ и управление риском (Animal tissues and their derivatives utilized in the manufacture of medical devices - Analysis and management of risk) |
[35] | БС ЕН 12442-2:2000 | Ткани животных и их производные, применяемые для изготовления медицинских изделий. Часть 2. Контроль получения, сбора и обращения (Animal tissues and their derivatives utilized in the manufacture of medical devices - Controls on sourcing, collection and handling) |
[36] | БС ЕН 12442-3:2000 | Ткани животных и их производные, применяемые для изготовления медицинских изделий. Часть 3. Валидация уничтожения и/или инактивации вирусов и переносимых возбудителей болезней (Animal tissues and their derivatives utilized in the manufacture of medical devices - Validation of the elimination and/or inactivation of viruses and transmissible agents) |
[37] | EH 13060:2004 | Стерилизаторы небольшие паровые (Small steam sterilizers) |
[38] | Good Automated Manufacturing Practice, Guide for Validation of Automated Systems in Pharmaceutical Manufacturing (GAMP 4) 2001. | |
[39] | Global Harmonization Task Force (GHTF) -Study Group 1 (SG1), Document No. N029R11, 2 Feb., 2002 | |
[40] | National Canners Association, Manual for Food Canners and Processors, Vol. 1, AVI Publishing Co., Westport, CT, 1968 | |
[41] | Parenteral Drug Association, Technical Report No. 1 Revised 2007, Validation of Moist Heat Sterilization Processes: Cycle Design, Development, Qualification, and Ongoing Control, PDA Bethesda, MD 2007 | |
[42] | Release on the IAPWS Industrial Formulation 1997 for the Thermodynamic Properties of Water and Steam, Erlangen, Germany, September 1997 (IAPWS-IF97) published in ASME. International Steam Tables for Industrial Use, ASME Press, New York NY 10016, 2000, ISBN 0-7918-01543 | |
[43] | Bowie, J.H., Kelsey, J.С and Thompson, G.R. The Bowie and Dick autoclave test, Lancet, pp.586-587, 1963 | |
[44] | Chen, J.H.S. Methods of testing virucides, Disinfection, Sterilization and Preservation, in Block S.S. (ed), Lea and Febinger, Philadelphia, PA | |
[45] | Halvorson, H.O. and Ziegler, N.R. Applications of statistics to problems in bacteriology, Journal of Bacteriology 25(2), pp.101-118, 1933 | |
[46] | Irvine, Th.F and Liley, P. E. Steam and Gas tables with computer equations, Academic Press, 1984 | |
[47] | Pflug, I.J. and Kristen, D.E. Carrying out Biological Qualification, the Control Operation of Moist-Heat (Steam Sterilization) Processes for Producing Sterile Pharmaceuticals and Medical Devices, PDA Journal of Pharmaceutical Science and Technology, Vol. 54(2) March/April 2000 | |
[48] | Pflug, I.J. Microbiology and Engineering of Sterilization Processes, St Paul University of Minnesota, Department of Food and Science and Nutrition and Schools of Public Health, 1992, Tenth Edition, 1999 | |
[49] | Morrissey, R.F. and Phillips, G.B. (eds.), Sterilization technology: A practical guide for manufacturers and users of health care products, Van Nostrant Reinhold, New York, 1995 | |
[50] | Parry, W.T., Bellows, J.C., Gallagher, J.S. and Harvey, A.H. Asme International Steam Tables for Industrial Use, ASME Press, New York, 2000, ISBN 0-7918-01543 | |
[51] | Pflug, I.J. and Hholcomb, R. G. Principles of thermal destruction of microorganisms, in Block, S.S. | |
[52] | Pflug, I.J. Microbiology and Engineering of Sterilization Processes (Микробиология и инжиниринг процессов стерилизации), 1992 | |
[53] | ИСО 11140-1 | Стерилизация медицинской продукции. Химические индикаторы. Часть 1. Общие требования (Sterilization of health care products - Chemical indicators - Part 1: General Requirements) |
[54] | ИСО 11140-3 | Стерилизация медицинской продукции. Химические индикаторы. Часть 3. Системы индикаторов класса 2 для испытания на паропроницание по методу Буи и Дика (Sterilization of health care products - Chemical indicators - Part 3: Class 2 indicator systems for use in the Bowie and Dick-type steam penetration test) |
[55] | ИСО 11140-4 | Стерилизация медицинской продукции. Химические индикаторы. Часть 4. Индикаторы класса 2 как альтернатива испытанию для обнаружения паропроницания по методу Буи и Дика (Sterilization of health care products - Chemical indicators - Part 4: Class 2 indicators as an alternative to the Bowie and Dick-type test for detection of steam penetration) |
[56] | ИСО 11140-5 | Стерилизация медицинской продукции. Химические индикаторы. Часть 5. Индикаторы класса 2 для испытания на эффективность удаления воздуха по методу Буи и Дика (Sterilization of health care products - Chemical indicators - Part 5: Class 2 indicators for Bowie and Dick-type air removal tests) |
[57] | ИСО 3746 | Акустика. Определение уровней звуковой мощности и уровней звуковой энергии источников шума по звуковому давлению. Ориентировочный метод с использованием охватывающей измерительной поверхности над звукоотражающей плоскостью (Acoustics - Determination of sound power levels and sound energy levels of noise sources using sound pressure - Survey method using an enveloping measurement surface over a reflecting plane) |
[58] | ANSI/AAMI ST79 | Comprehensive guide to steam sterilization and sterility assurance in health care facilities |
УДК 615.478.73:006.354 | ОКС 11.080.01 | Р26 | ОКП 94 5120 |
Ключевые слова: стерилизация, влажное тепло, валидация, текущий контроль | |||
Электронный текст документа
и сверен по:
, 2016

{kind=link}